
Β. Δαράκη
Επιμελητρια Β’ ΕΣΥ, Κλινική Ενδοκρινολογίας, Διαβήτη και Μεταβολικών Νοσημάτων,
Πανεπιστημιακό Νοσοκομείο Ηρακλείου
Ε. Μαμαλάκη
Διευθύντρια ΕΣΥ, Κλινική Ενδοκρινολογίας, Διαβήτη και Μεταβολικών Νοσημάτων,
Πανεπιστημιακό Νοσοκομείο Ηρακλείου
Εισαγωγή
Η πρόοδος της μοριακής βιολογίας επέτρεψε τα τελευταία χρόνια την καλύτερη κατανόηση των μηχανισμών του κυτταρικού πολλαπλασιασμού και της ρύθμισης του κυτταρικού κύκλου, του προγραμματισμένου κυτταρικού θανάτου (απόπτωση), της αγγειογένεσης, της διήθησης και τέλος της μετάστασης.
Η καρκινογένεση αποτελεί μία πολυπαραγοντική διαδικασία όπου γενετικές αλλαγές και καρκινογόνα (π.χ χημικοί παράγοντες, περιβαλλοντικές επιδράσεις, ιοί κ.α) τροποποιούν τη αλληλουχία του DNA γονιδίων υπεύθυνων για τα ανωτέρω μόρια (ογκογονίδια) με συνέπεια τον ανεξέλεγκτο κυτταρικό πολλαπλασιασμό (1). Πολλά ογκογονίδια σχετίζονται εκτός από την καρκινογένεση με τον τύπο του καρκίνου, την ικανότητα διαφοροποίησης, την πρόγνωση και σε μερικές περιπτώσεις την ανταπόκρισή σε ειδικές θεραπείες.
Ο καρκίνος του θυρεοειδούς αποτελεί τη πιο συχνή νεοπλασία του ενδοκρινικού συστήματος με συνεχώς αυξανόμενη επίπτωση τις τελευταίες δεκαετίες (2). Η μοριακή βάση της καρκινογένεσης στον θυρεοειδή έχει μελετηθεί εκτεταταμένα τα τελευταία χρόνια. Σε αυτήν φαίνεται ότι εμπλέκονται α) πρωτοογκογονίδια (proto-oncogenes) τα προϊόντα των οποίων σχετίζονται με την αύξηση ή την διαίρεση του κυττάρου β) ογκοκατασταλτικά γονίδια(tumor suppressor genes) τα προϊόντα των οποίων σχετίζονται με την αναστολή της αύξησης και του πολλαπλασιασμού του κυττάρου και γ) γονίδια-μεταλλάκτες τα προϊόντα των οποίων εμπλέκονται στην αντιγραφή και στην επιδιόρθωση του DNA. Οι μεταλλάξεις στα ογκοκατασταλτικά γονίδια είναι υπολειπόμενες και δεν αρκούν από μόνες τους να οδηγήσουν σε καρκινογένεση. Αντίθετα στα πρωτοογκογονίδια η μετάλλαξη ενός μόνο αλληλομόρφου αρκεί για να οδηγήσει σε ανεξέλεγκτο κυτταρικό πολλαπλασιασμό. Αξίζει να αναφερθεί ότι τα πρωτοογκογονίδια είναι δυνατόν να υποστούν αναδιάταξη του DNA τους και συγχώνευση με τμήματα άλλων γονιδίων (χιμαιρικά ογκογονίδια) που εξυπηρετούν βασικές κυτταρικές λειτουργίες. Το αποτέλεσμα είναι η συνεχής ενεργοποίησή τους και η συνεχής αύξηση και πολλαπλασιασμός του κυττάρου το οποίο εξελίσσεται σε νεοπλασματικό. Στην Εικόνα 1 φαίνεται η αλληλουχία των γενετικών αλλαγών που οδηγεί το φυσιολογικό θυρεοειδικό κύτταρο σε καλοήθη υπερπλασία/ανάπτυξη αδενώματος, είτε σε ανάπτυξη καλά διαφοροποιημένου καρκίνου θυρεοειδούς που μπορεί να εξελιχθεί σε νεόπλασμα φτωχής διαφοροποίησης και τελικά σε αναπλαστικό καρκίνωμα. Οι ανωτέρω μεταλλάξεις και αναδιατάξεις γονιδίων χρησιμοποιούνται σήμερα ως μοριακοί δείκτες στον καρκίνο του θυρεοειδούς. Επίσης, ως μοριακοί δείκτες χρησιμοποιούνται και δείκτες βασιζόμενοι στο RNA καθώς και ανοσοιστοχημικοί δείκτες στον ορό των ασθενών.

Εικόνα 1. Moντέλο της καρκινογένεσης στον θυρεοειδή. Αυτό το μοντέλο βασίζεται σε ιστολογικά και κλινικά γνωρίσματα καθώς και στο βαθμό διαφοροποίησης του καρκίνου του θυρεοειδούς. Το θυλακικό κύτταρο μπορεί να εξελιχθεί σε καλόηθες αδένωμα είτε σε νεοπλασματικό όγκο. Αυτόνομα υπερλειτουργούντα αδενώματα θυρεοειδούς συνδέονται με ενεργοποιητικές μεταλλάξεις του υποδοχέα της TSH (TSHR) ή των G-πρωτεϊνών(3). Εξέλιξη του θυλακικού κυττάρου σε νεοπλασματικό οφείλεται σε ενεργοποιητικές μεταλλάξεις διαφορετικών πρωτοογκογονιδίων και ογκοκατασταλτικών γονιδίων, με αποτέλεσμα την ανάπτυξη θηλώδους και θυλακιώδους καλά διαφοροποιημένου καρκίνου θυρεοειδούς, καρκίνου χαμηλής διαφοροποίησης και αναπλαστικού καρκινώματος. Πηγή: (4).
1. Μεταλλάξεις γονιδίων και αναδιατάξεις
1.1. Θηλώδες καρκίνωμα θυρεοειδούς
Το θηλώδες καρκίνωμα αποτελεί τον πιο συχνό τύπο καρκίνου θυρεοειδούς και αναλογεί στο 80% περίπου των περιπτώσεων (5). Ένα υψηλό ποσοστό (70%) θηλωδών νεοπλασμάτων οφείλονται σε γενετικές διαταραχές που οδηγούν σε ενεργοποίηση του μονοπατιού μετάδοσης σήματος της MAPK κινάσης (6). Αυτές περιλαμβάνουν σημειακές μεταλλάξεις των γονιδίων BRAF και RAS καθώς και χιμαιρικά ογκογονίδια, κυρίως το RET/PTC και σπανιότερα το TRK1 (7,8,9) καθένα από τα οποία μπορούν να ενεργοποιήσουν ξεχωριστά το ανωτέρω μονοπάτι (Εικόνα 2). Οι πιο συχνές μεταλλάξεις που οδηγούν στην ανάπτυξη θηλώδους καρκινώματος είναι μεταλλάξεις του γονιδίου BRAF (45%) και ακολουθούν μεταλλάξεις του RAS (15%) και του χειμερικού ογκογονιδίου RET/PTC (15%) (10). Η επίδραση των μεταλλάξεων του BRAF και του RET/PTC στον κοινό μονοπάτι μετάδοσης σήματος φαίνεται ότι διαφέρει καθώς οδηγούν σε διαφορετικούς υποτύπους θηλώδους καρκίνου θυρεοειδούς (11).
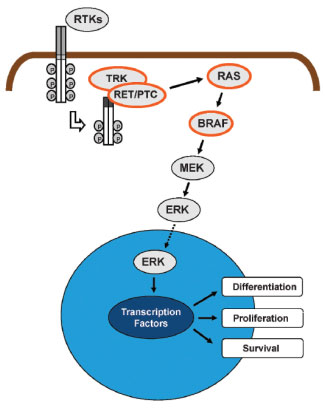
Εικόνα 2. Όλες οι μεταλλάξεις που ανευρίσκονται στα θηλώδη καρκινώματα θυρεοειδούς είναι δυνατόν να ενεργοποιήσουν το μονοπάτι σηματοδότησης της MAPK κινάσης. Αυτό το μονοπάτι μεταφέρει μηνύματα από υποδοχέα τυροσινικών κινασών της κυτταρικής μεμβράνης (RTKs) στον πυρήνα διαμέσου μιας σειράς πρωτεϊνών και ενδοκυτταροπλασματικών κινασών περιλαμβανομένων των RAS, RAF (κυρίως το BRAF στα θυλακιώδη κύτταρα του θυρεοειδούς), MEK, και ERK. Το τελευταίο όταν ενεργοποιηθεί ρυθμίζει την μεταγραφή γονιδίων στον πυρήνα που εμπλέκονται στην διαφοροποίηση, στον πολλαπλασιασμό και στην επιβίωση του κυττάρου. Πηγή: (9).
1.1.1. Χιμαιρικό ογκογονίδιο RET/PTC
Το ογκογονίδιο RET βρίσκεται στο χρωμόσωμα 10 και κωδικοποιεί έναν διαμεμβρανικό υποδοχέα κινάσης της τυροσίνης, που ανήκει στην οικογένεια του υποδοχέα του αυξητικού παράγοντα των αιμοπεταλίων (PDGF-R) (12). Υπό φυσιολογικές συνθήκες ενεργοποιείται από τέσσερεις διαφορετικούς παράγοντες: GDNF (glial cell derived neurotrophic factor), Neurturin (NRTN), Artimin (ARTN), Persepin (PSPN) (13,14) και φαίνεται ότι συμμετέχει στην ανάπτυξη του περιφερικού νευρικού συστήματος κατά την ενδομήτριο ζωή (15). Το γονίδιο RET εκφράζεται φυσιολογικά σε ιστούς που προέρχονται από τη νευρική ακρολοφία όπως τα κύτταρα C του θυρεοειδούς και ο μυελός των επινεφριδίων, όχι όμως στα κύτταρα των θυλακίων του θυρεοειδούς (16).
Το 1987 ανακαλύφτηκε για πρώτη φορά το χιμαιρικό ογκογονίδιο RET/PTC σε κύτταρα θηλώδους καρκινώματος θυρεοειδούς (17). Η κλωνοποίησή του 3 χρόνια αργότερα έδειξε ότι προέρχεται από τη συγχώνευση περιοχής του ογκογονιδίου RET που φέρει την ιδιότητα της τυροσινικής κινάσης με τη ρυθμιστική περιοχή του γονιδίου CCD6 (Εικόνα 3) (18). Σήμερα έχουν περιγραφεί 13 διαφορετικοί τύποι RET/PTC αναδιατάξεων. Τα γονίδια που συγχωνεύονται με τμήμα του RET ογκογονιδίου βρίσκονται στο ίδιο χρωμόσωμα με το RET και έχουν υψηλό επίπεδο έκφρασης στα κύτταρα του θυρεοειδούς κωδικοποιώντας πρωτεΐνες που εξυπηρετούν βασικές λειτουργίες του κυττάρου. Το αποτέλεσμα είναι να παράγεται διαρκώς η χιμαιρική πρωτεΐνη RET/PTC και να προκαλείται συνεχής ενεργοποίηση του μονοπατιού RAS-RAF-MAPK που οδηγεί στον ανεξέλεγκτο πολλαπλασιασμό και στην ανάπτυξη νεοπλασίας. Κύριο αίτιο της δημιουργίας των ανωτέρω αναδιατάξεων είναι η έκθεση σε ακτινοβολία που προκαλεί βλάβες στο DNA του κυττάρου, όπως αποδείχτηκε σε in vitro πειράματα (19, 20). Ωστόσο τα τελευταία έτη έχει περιγραφεί ο ρόλος και άλλων πιθανών καρκινογόνων όπως η καφεΐνη, η αιθανόλη η υποξία κ.α. (21) καθώς και παραγόντων που αυξάνουν τη συγκέντρωση ανιόντων υπεροξειδίου και H2O2 (22) στην εμφάνιση αναδιατάξεων RET/PTC.
Το RET/PTC1 είναι η πιο συχνή αναδιάταξη στα θηλώδη καρκινώματα (60-70% των αναδιατάξεων). Έχει περιγραφεί σε ποσοστό 10-20% σποραδικών θηλωδών νεοπλασμάτων θυρεοειδούς αν και ο επιπολασμος τους ποικίλει σημαντικά σε διάφορες μελέτες (23) και είναι ιδιαίτερα χαμηλός σε θηλώδη με θυλακιώδη διαμόρφωση (24). Η ανεύρεσή τους σε μικροκαρκινώματα ενισχύει την υπόθεση ότι πρόκειται για γενετική βλάβη που συμβαίνει πρώιμα στη διαδικασία της καρκινογένεσης (25). Η επίπτωσή τους είναι υψηλότερη σε ασθενείς με ιστορικό έκθεσης σε ακτινοβολία (50-80%) όπως σε επιβιώσαντες από ατομική βόμβα (26) και σε ασθενείς με καρκίνο θυρεοοειδή μετά το πυρηνικό ατύχημα του Chernobil (27). Επίσης είναι υψηλότερη σε παιδιά και νέους ενήλικες, ανεξάρτητα από την έκθεση σε ακτινοβολία (40-70%) (28,29). Αν και σχετίζεται με υψηλό ποσοστό λεμφαδενικών μεταστάσεων (34), φαίνεται ότι έχει καλή πρόγνωση (35). Eπίσης ανευρίσκεται σε νεοπλάσματα Hurthle με χαρακτηριστικά θηλωδών νεοπλασιών θυρεοειδούς (36) καθώς και στους ιστολογικούς υποτύπους θηλώδους με επιθετικό φαινότυπο (sclerosing και tall-cell variant (37) στους ενήλικες και solid variant στα παιδιά (28).
Η κατανομή της αναδιάταξης RET/PTC μπορεί να αφορά τα περισσότερα νεοπλασματικά κύτταρα (clonal RET/PTC) του όγκου -ειδική για το θηλώδες νεόπλασμα του θυρεοειδούς (23,9) ή να ανιχνεύεται σε μικρό ποσοστό (<1%) των νεοπλασματικών κυττάρων(nonclonal RET/PTC) (30,31). Η τελευταία έχει βρεθεί εκτός από θηλώδη καρκινώματα σε αδενώματα θυρεοειδούς και άλλες καλοήθεις βλάβες (32) καθώς και σε θυρεοειδίτιδα Hashimoto (33).
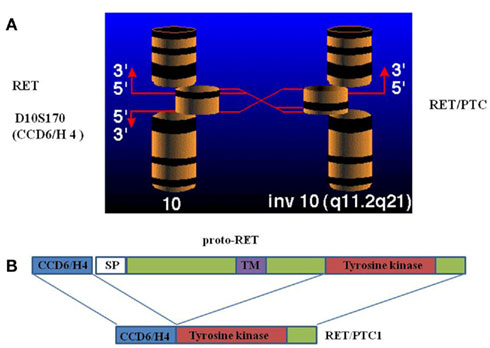
Εικόνα 3. Σχηματική αναπαράσταση της αναδιάταξης του γονιδίου RET με την περιοχή 5΄άλλου γονιδίου που εκφράζεται στον θυρεοειδή. Πηγή: (12).
1.1.2. Χιμαιρικό oγκογονίδιο TRK
Το γονίδιο NTRK1 κωδικοποιεί τον υποδοχέα για τον αυξητικό παράγοντα των νεύρων Trk-A. Ο υποδοχέας αυτός έχει ιδιότητες τυροσινικής κινάσης και όταν ενεργοποιηθεί αυτοφωσφορυλιώνεται και ταυτόχρονα φωσφορυλιώνει πρωτεΐνες από το μονοπάτι της MAPK κινάσης. Παίζει σημαντικό ρόλο στην ανάπτυξη και ωρίμανση του κεντρικού και περιφερικού νευρικού συστήματος και επιπρόσθετα επάγει τον πολλαπλασιασμό μεγάλου αριθμού κυττάρων όπως τα λεμφοκύτταρα και τα κύτταρα του προστάτη (34,35,36).
Η αλληλουχία των νουκλεοτιδίων του NTRK1 που κωδικοποιεί την περιοχή με ιδιότητα κινάσης της τυροσίνης του υποδοχέα αυτού, μπορεί να ενωθεί με αλληλουχίες νουκλεοτιδίων άλλων γονιδίων δημιουργώντας τα χιμαιρικά ογκογονίδια TRK, TRK-T1, TRK-T2, TRK-T3. Τα ανωτέρω ανευρίσκονται στο θηλώδες καρκίνωμα θυρεοειδούς σε ποσοστό 12% (με ποιο συχνό το TRK), και δεν φαίνεται να σχετίζονται με την έκθεση στην ακτινοβολία (37,38). Μελέτες σε περιορισμένο αριθμό ασθενών συσχετίζουν την ύπαρξη των χειμερικών ογκογονιδίων TRK με την εμφάνιση θηλώδους καρκίνου θυρεοειδούς σε νεαρή ηλικία και με χειρότερη πρόγνωση της νόσου (39,40).
Επίσης πειράματα σε ζώα δείχνουν ότι τα χειμερικά ογκογονίδια TRK αποτελούν πρώιμο γεγονός στην καρκινογένεση των κυττάρων του θυρεοειδούς, για την επαγωγή της οποίας χρειάζονται τη συνύπαρξη ογκοκατασταλτικών ογκογονιδίων (41).
1.1.3. Πρωτο-ογκογονίδιο Ras
Τα γονίδια RAS (HRAS, KRAS and NRAS) κωδικοποιούν μικρές ενδοκυττάριες GTPάσες με καθοριστικό ρόλο στη μεταφορά των ενδοκυττάριων σημάτων από διαμεμβρανικούς υποδοχείς (42). Η μη ενεργοποιημένη RAS πρωτεΐνη είναι συνδεδεμένη με το GDP. Μετά την ενεργοποίησή της απελευθερώνει GDP και συνδέεται με GTP ενεργοποιώντας το μονοπάτι της MAPK κινάσης αλλά και άλλα σημαντικά μονοπάτια όπως το PI3K/AKT (Εικόνα 4). Φυσιολογικά η ενεργοποιημένη RAS-GTP πρωτεϊνη απενεργοποιείται γρήγορα (43). Σημειακές μεταλλάξεις στο γονίδιο RAS είτε αυξάνουν την ικανότητα σύνδεσης με το GTP (μεταλλάξεις στα κωδικόνια 12 and 13), είτε απενεργοποιούν την αυτοκαταλυτική δράση ως GTPase (μεταλλάξεις στο κωδικόνιο 61) (44). Ως αποτέλεσμα, η μεταλλαγμένη πρωτεΐνη παραμένει διαρκώς ενεργοποιημένη και προκαλεί συνεχή σηματοδότηση μέσα στο κύτταρο των MAPK και PI3K/AKT μονοπατιών παρά την απουσία εξωτερικών ερεθισμάτων, οδηγώντας σε συνεχή πολλαπλασιασμό του κυττάρου (42). Το πρωτο-ογκογονίδιο Ras είναι το πιο συχνό ογκογονίδιο στον καρκίνο του ανθρώπου. Μεταλλάξεις του ανευρίσκονται σε 20-25% όλων των καρκίνων και πάνω από 90% σε συγκεκριμένους τύπους καρκίνου (45).
Όσον αφορά τον καρκίνο του θυρεοειδή έχουν βρεθεί μεταλλάξεις που αφορούν και τις τρεις πρωτεΐνες RAS με πιο συχνές τις μεταλλάξεις στα γονίδια NRAS και HRAS (43) ωστόσο ο ρόλος τους στην εξέλιξη των καρκινικών κυττάρων δεν είναι σαφής.
Στα θηλώδη καρκινώματα μεταλλάξεις στο γονίδιο RAS (κυρίως στο Κ-RAS) συμβαίνουν στο 10-20% των όγκων (44,46,47). Αφορούν σχεδόν αποκλειστικά τα θηλώδη καρκινώματα με θυλακιώδη διαμόρφωση που περιβάλλονται εξ ολοκλήρου ή ατελώς από κάψα και σχετίζονται με χαμηλή συχνότητα λεμφαδενικών μεταστάσεων και καλύτερη πρόγνωση (48,49). Ωστόσο μεταλλάξεις του Ν-RAS στο κωδικόνιο 61 ενοχοποιούνται για δυσμενή εξέλιξη στα θηλώδη καρκινώματα και αποτελούν ανεξάρτητο προγνωστικό παράγοντα (50).
1.1.4. Πρωτο-ογκογονίδιο BRAF
Το πρωτο-ογκονίδιο BRAF κωδικοποιεί την πρωτεϊνική κινάση B-RAF της σερίνης/θρεονίνης μέλος της οικογένειας των RAF κινασών, που συμμετέχουν στην ενδοκυττάρια μεταφορά μηνυμάτων (9). Η συγκεκριμένη πρωτεΐνη δέχεται το μιτογόνο σήμα από την πρωτεΐνη RAS και το προωθεί προς την οδό ενεργοποίησης της ΜΑP κινάσης. Κληρονομούμενες μεταλλάξεις του γονιδίου BRAF προκαλούν σύνδρομο που χαρακτηρίζεται από χαρακτηριστικό προσωπείο, ανωμαλίες της καρδιάς και διανοητική καθυστέρηση (51). Επίκτητες μεταλλάξεις αυτού του γονιδίου έχουν βρεθεί σε αρκετές μορφές καρκίνου όπως το non-Hodgkin λέμφωμα, ο καρκίνος του παχέος εντέρου, το κακόηθες μελάνωμα, το μη-μικροκυτταρικό καρκίνωμα του πνεύμονα, και το αδενοκαρκίνωμα του πνεύμονα (52).
Όσον αφορά τον θυρεοειδή, σημειακές μεταλλάξεις του γονιδίου BRAF αποτελούν τις πιο συχνές γενετικές διαταραχές στα θηλώδη καρκινώματα και ανευρίσκονται στο 45% των όγκων αυτών. Η συνηθέστερη μετάλλαξη είναι η V600E η οποία οδηγεί σε συνεχή φωσφορυλίωση της MEK και ενεργοποίηση του μονοπατιού της MAPK κινάσης (Εικόνα 4) (53). Ανευρίσκεται με υψηλή συχνότητα στα θηλώδη καρκινώματα με τον κλασσικό ιστολογικό τύπο καθώς και σε επιθετικούς υπότυπους (υποτύπος με υψηλά κύτταρα (tall-cell variant), διηθητκό θηλώδες με θυλακιώδη διαμόρφωση) (54) αλλά είναι σπάνιο στα θηλώδη καρκινώματα με θυλακιώδη διαμόρφωση που περιβάλλονται από κάψα (55).
Επίσης ανιχνεύεται σε φτωχής διαφοροποίησης καθώς και σε αναπλαστικά νεοπλάσματα θυρεοειδούς που προέρχονται από καλά διαφοροποιημένο θηλώδες καρκίνωμα. Η ανεύρεση της τόσο στα καλά διαφοροποιημένα τμήματα όσο και στα τμήματα φτωχής διαφοροποίησης του ίδιου όγκου υποδηλώνει ότι συμβαίνει νωρίς στην διαδικασία καρκινογένεσης στον θυρεοειδή (56). Το γεγονός ότι ανιχνεύεται σπάνια σε θυλακιώδη καρκινώματα ή καλοήθη αδενώματα θυρεοειδούς την καθιστά αρκετά ειδικό δείκτη διάγνωσης θηλώδους καρκινώματος και νεοπλασμάτων προερχόμενων από αυτό (56).
Άλλες μεταλλάξεις του γονιδίου BRAF ανευρίσκονται σε 1-2% θηλωδών καρκινωμάτων. Τέτοιες είναι η K601E σημειακή μετάλλαξη, που έχει περιγραφεί και σε μία περίπτωση θυλακιώδους αδενώματος (57), μικρές προσθήκες ή απαλοιφές γύρω από το κωδικόνιο 600 και η αναδιάταξη AKAP9-BRAF που είναι πιο συχνή σε θηλώδη καρκινώματα που σχετίζονται με έκθεση σε ακτινοβολία (58).
Σε πολλές μελέτες η παρουσία μεταλλάξεων του BRAF γονιδίου σχετίζεται με επιθετικά χαρακτηριστικά του όγκου, όπως εξωθυρεοειδική επέκταση, προχωρημένο στάδιο κατά τη διάγνωση, υποτροπή, λεμφαδενικές ή απομακρυσμένες μεταστάσεις (59). Ειδικά η μετάλλαξη BRAF V600E έχει βρεθεί ότι είναι ανεξάρτητος προγνωστικός παράγοντας υποτροπής του όγκου, ακόμη και σε ασθενείς σε στάδιο Ι-ΙΙ της νόσου (59,60). Επίσης η μετάλλαξη αυτή έχει συσχετισθεί με μειωμένη ικανότητα των όγκων να προσλαμβάνουν ραδιενεργό ιώδιο καθώς και αποτυχία αντιμετώπισης υποτροπής της νόσου, που μπορεί να οφείλεται σε απορρύθμιση της λειτουργίας του συμμεταφορέα νατρίου-ιωδίου (NIS) και άλλων γονιδίων που συμμετέχουν στον μεταβολισμό του ιωδίου στα θυλακιώδη κύτταρα του θυρεοειδούς (61).
Η συμμετοχή μεταλλάξεων του γονιδίου BRAF στην έναρξη της καρκινογένεσης στον θυρεοειδή και στην αποδιαφοροποίηση των κυττάρων καθώς και η συσχέτισή τους με πιο επιθετικά χαρακτηριστικά των όγκων έχει φανεί και σε μελέτες σε ποντίκια που έφεραν την ειδική για τον θυρεοειδή έκφραση της V600E BRAF μετάλλαξης (62).
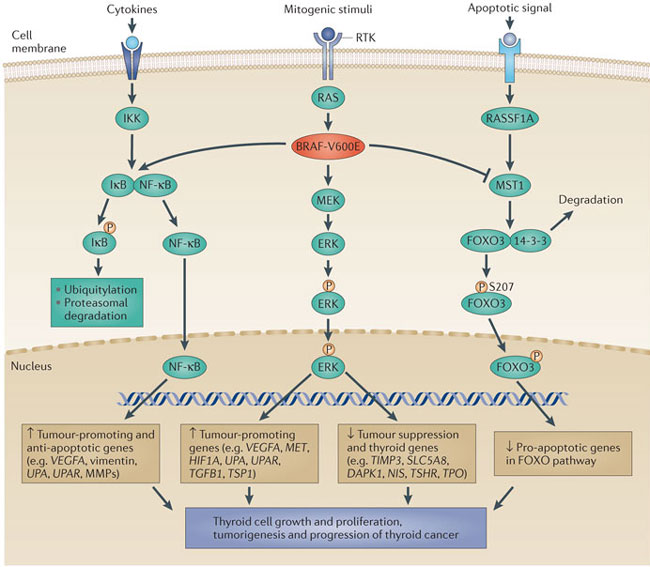
Εικόνα 4. Η μετάλλαξη BRAF-V600E αποτελεί καθοριστικό παράγοντα για την εξέλιξη της καρκινογένεσης στον θυρεοειδή. Πηγή: (63).
1.1.5. Πρώτο-ογκογονίδιο met
Το πρωτο-ογκογονίδιο met κωδικοποιεί έναν υποδοχέα για τον ηπατοκυτταρικό αυξητικό (HGF/SF) με δράση τυροσινικής κινάσης (64). Ο υποδοχέας αυτός φυσιολογικά παίζει καθοριστικό ρόλο στην εμβρυογένεση (65), αλλά και στην αναγέννηση του ήπατος καθώς και στην επούλωση πληγών κατά την ενήλικο ζωή (66). Η δέσμευσή του ενεργοποιεί πολλαπλά μονοπάτια μετάδοσης σήματος (μονοπάτι RAS (67), μονοπάτι PI3K (68), μονοπάτι STAT (69), μονοπάτι beta-catenin (70), μονοπάτι Νotch (71), με αποτέλεσμα μεταλλάξεις που το ενεργοποιούν μόνιμα να οδηγούν σε μιτογένεση και να προάγουν την κινητικότητα του κυττάρου και την διήθηση (72). Η έκφραση του πρωτο-ογκογονιδίου c-Met είναι ένας σημαντικός δείκτης για το θηλώδες καρκίνωμα θυρεοειδούς από υψηλά κύτταρα και τη διηθητική του συμπεριφορά και ενεδομένως έχει ρόλο στην έγκαιρη αναγνώριση ασθενών που πάσχουν από αυτό (73).
1.1.6. Ο υποδοχέας του επιθηλιακού αυξητικού παράγοντα (EGFR)
Ο EGFR ανήκει στην οικογένεια των υποδοχέων τυροσινικής κινάσης. Έχει αυξημένη έκφραση στο θηλώδες καρκίνωμα συγκριτικά με τα φυσιολογικά κύτταρα θυρεοειδούς. Σε μία μελέτη όπου διερευνήθηκε η έκφραση των τεσσάρων γονιδίων του υποδοχέα αυτού στον καλά διαφοροποιημένο καρκίνο του θυρεοειδούς βρέθηκε ότι τουλάχιστον ένα υπερεκφραζόταν και ότι μόνο το HER3 σχετιζόταν με το στάδιο της νόσου (74). Αν και μέχρι πρότινος δεν θεωρούνταν σημαντικός στη μοριακή βιολογία του καρκίνου του θυρεοειδούς, πρόσφατη μελέτη έδειξε ότι EGFR μεταλλάξεις που ανευρίσκονται σε καρκίνο του πνεύμονα μπορούν να ανευρεθούν και σε ένα υποσύνολο θηλώδους ή φτωχής διαφοροποίησης καρκίνου θυρεοειδούς καθιστώντας τα καρκινικά κύτταρα ευαίσθητα σε αναστολείς του συγκεκριμένου ογκογονιδίου (75).
1.1.7. Ο αγγειακός επιθηλιακός αυξητικός παράγοντας (VEGF) και ο υποδοχέας του
Η υπερέκφραση του VEGF είναι χαρακτηριστικό εύρημα νεοπλασιών συμπεριλαμβανομένων και του καρκίνου του θυρεοειδούς και μπορεί να χαρακτηρισθεί ως δείκτης υποξίας του όγκου (76). Η έκφραση του VEGF σχετίζεται με μεταστατικό θηλώδες καρκίνωμα θυρεοειδούς και συγκεκριμένα με το μέγεθος του όγκου, με εξωθυρεοειδική επέκταση και με την παρουσία μεταλλάξεων του BRAF ογκογονιδίου (77), καθώς και με την ύπαρξη λεμφαδενικών μεταστάσεων (78).
1.2. Θυλακιώδες καρκίνωμα θυρεοειδούς
Τα θυλακιώδη καρκινώματα του θυρεοειδούς αναλογούν στο 10-15% των νεοπλασιών του θυρεοειδούς. Είναι συνήθως μονοεστιακά. Αντίθετα με τα θηλώδη νεοπλάσματα οι απομακρυσμένες μεταστάσεις κυρίως στον πνεύμονα και στα οστά είναι συχνές κατά τη διάγνωση τους, ενώ οι λεμφαδενικές μεταστάσεις είναι σπανιότερες (< 5%) (5).
Στην ανάπτυξη θυλακιώδους καρκινώματος του θυρεοειδούς εκτός από μεταλλάξεις του ογκογονιδίου RAS (20-40%), εμπλέκονται αναδιατάξεις PAX8-PPARγ (30-40%), καθώς και γενετικές αλλαγές που αφορούν το μονοπάτι μετάδοσης σήματος PI3K/AKT σε χαμηλότερο ποσοστό (15%). Οι τελευταίες φαίνεται ότι παίζουν ρόλο στα τελευταία στάδια ανάπτυξης του όγκου και ότι εμπλέκονται μαζί με μεταλλάξεις των γονιδίων the TP53 and CTNNB1, με άλλοτε άλλη συχνότητα στην ανάπτυξη νεοπλασμάτων φτωχής διαφοροποίησης και αναπλαστικών καρκινωμάτων (3). Πολλές από τις ανωτέρω μεταλλάξεις συνδέονται με διαφορετικά φαινοτυπικά χαρακτηριστικά των όγκων και μερικές αποτελούν δείκτες πιο επιθετικής συμπεριφοράς. Πρόσφατες μελέτες δείχνουν ότι η συνύπαρξη πολλαπλών γενετικών διαταραχών σε ένα ασθενή σχετίζεται με την εμφάνιση καρκίνου θυρεοειδούς σε νεαρότερη ηλικία (3).
1.2.1. Χιμαιρικό ογκογονίδιο PAX8/PPARγ
Το χιμαιρικό ογκογονίδιο PAX8/PPARγ προκύπτει από την αναδιάταξη μεταξύ των χρωμοσωμάτων 2 και 3, t(2;3)(q13;p25) που οδηγεί σε συγχώνευση τμήματος του γονιδίου του μεταγραφικού παράγοντα PAX8 και τμήματος του γονιδίου του μεταγραφικού παράγοντα PPARγ (Εικόνα 5). Ο παράγοντας PAX8 εμπλέκεται στην ανάπτυξη και διαφοροποίηση των θυλακιωδών κυττάρων του θυρεοειδούς (79), ενώ ο παράγοντας PPARγ ανήκει στους πυρηνικούς υποδοχείς και έχει αμφίβολο ρόλο στην καρκινογένεση (80). Η αναδιάταξη PAX8/PPARγ οδηγεί σε υπερέκφραση της πρωτεΐνης PPARγ.
Το χιμαιρικό ογκογονίδιο PAX8/PPARγ ανευρίσκεται στο 30-40% των περιπτώσεων θυλακιώδους καρκίνου θυρεοειδούς, ενώ ο επιπολασμός του στις περιπτώσεις ογκοκυτταρικού καρκίνου (κύτταρα Hurthe) είναι χαμηλός (81,82,83). Οι όγκοι που φέρουν αυτήν την αναδιάταξη τείνουν να εμφανίζονται σε νεαρότερη ηλικία, είναι μικρότεροι σε μέγεθος και συχνά παρουσιάζουν αγγειακή διήθηση (82,81)
Το ογκογονίδιο PAX8/PPARγ ανευρίσκεται επίσης σε μικρό ποσοστό (2-10%) σε θυλακιώδη αδενώματα και σε ποσοστό 0.5% σε θηλώδες καρκίνο θυρεοειδούς με θυλακιώδη διαμόρφωση (81,84,85). Τα θυλακιώδη αδενώματα θετικά γι αυτήν την αναδιάταξη έχουν παχιά κάψα και εμφανίζουν ανοσοιστοχημικά χαρακτηριστικά καρκίνου θυρεοειδούς, οδηγώντας στην υπόθεση ότι πιθανόν αντιπροσωπεύουν in situ θυλακιώδη καρκινώματα ή νεοπλασίες όπου η διήθηση δεν φάνηκε αρχικά στην ιστολογική εξέταση. Γι αυτό η ανεύρεση του χιμαιρικού ογκογονιδίου PAX8/PPARγ σε χειρουργικά παρασκευάσματα οδηγεί τον παθολογοανατόμο να αναζητήσει με περισσότερη προσοχή δήθηση της κάψας ή των αγγείων εξετάζοντας ολόκληρη την κάψα του όγκου σε πολλαπλές ιστολογικές τομές (81,82).

Εικόνα 5. Σχηματικό διάγραμμα της αναδιάταξης μεταξύ του PAX8 στο χρωμόσωμα 2q13 και του PPARγ στο χρωμόσωμα 3p25. Πηγή: (86).
1.2.2. Πρωτο-ογκογονίδιο RAS
Αρχικές μελέτες έδειξαν παρόμοιο επιπολασμό σε μεταλλάξεις του RAS πρωτο-ογκογονιδίου τόσο σε καλοήθη θυλακιώδη αδενώματα, όσο και σε νεοπλασίες του θυρεοειδούς (87) υποδεικνύοντας ότι η ενεργοποίηση των RAS πρωτεϊνών μπορεί να αποτελεί ένα πρώιμο γεγονός στη διαταραχή πολλαπλασιασμού του κυττάρου του θυρεοειδούς (88,46). Μεταγενέστερες μελέτες έδειξαν ότι μεταλλάξεις ειδικά στο κωδικόνιο 61 του Ν-RAS εμπλέκονται στην περαιτέρω εξέλιξη σε νεοπλάσματα θυρεοειδούς με επιθετική κλινική συμπεριφορά (89,90,91).
Σε θυλακιώδη αδενώματα του θυρεοειδούς μεταλλάξεις του ογκογονιδίου RAS συμβαίνουν σε ποσοστό 20-40% και είναι πιο συχνές σε όσα έχουν μικροθυλακιώδες πρότυπο ανάπτυξης (87). Τα αδενώματα αυτά δεν έχουν μορφολογικά χαρακτηριστικά κακοήθειας (81). Στα θυλακιώδη καρκινώματα οι ανωτέρω μεταλλάξεις ανευρίσκονται σε ποσοστό 40-50% και αφορούν κυρίως μεταλλάξεις στο κωδικόνιο 61 του Ν-RAS (89). Χαμηλότερη επίπτωση των ανωτέρω μεταλλάξεων έχει βρεθεί σε όγκους από κύτταρα Hurthle όπου ανευρίσκονται μόνο σε 0-4% των αδενωμάτων και 15-25% των καρκινωμάτων (92, 93) .
1.2.3. Μεταλλάξεις πρωτεϊνών του μονοπατιού PI3K/AKT
Το κυτταρικό μονοπάτι PTEN/PI3K/Akt ρυθμίζει σημαντικές λειτουργίες του κυττάρου όπως το μεταβολισμό της γλυκόζης, τον κύκλο πολλαπλασιασμού και την επιβίωση του κυττάρου. Το γονίδιο PTEN κωδικοποιεί μία φωσφατάση που καταστέλλει το ανωτέρω μονοπάτι (Εικόνα 6) (94). Απενεργοποιητικές μεταλλάξεις του προκαλούν το σύνδρομο Cowden που χαρακτηρίζεται από αμαρτώματα του δέρματος και αδενώματα του εντέρου, του μαστού και του θυρεοειδούς και από αυξημένο κίνδυνο ανάπτυξης καρκίνου του μαστού, του θυρεοειδούς και του ενδομητρίου (95). Καλοήθη θυλακιώδη αδενώματα και καρκινώματα θυρεοειδούς εμφανίζονται σε 50-75% των ασθενών που πάσχουν από αυτό το σύνδρομο ενώ ο κίνδυνος εμφάνισης καρκίνου θυρεοειδούς κατά τη διάρκεια της ζωής των ασθενών κυμαίνεται σε 10% (96). Απενεργοποίηση του PTEN παίζει ρόλο και σε σποραδικά νεοπλάσματα θυρεοειδούς σπανιότερα στα θηλώδη (2%), συχνότερα στα θυλακιώδη (7%) και πολύ συχνά στα αμετάπλαστα (12-50%) (97,98,99).

Εικόνα 6. Το κλασσικό μονοπάτι PTEN Η ένωση του προσδέτη με μεμβρανικούς υποδοχείς ενεργοποιεί το PI3K. Το ενεργοποιημένο PI3K φωσφορυλιώνει το PI(3,4)P (PIP2) για να παραχθεί το PI(3,4,5)P (PIP3). Το τελικό αποτέλεσμα είναι η φωσφορυλίωση και ενεργοποίηση της AKT, η οποία ρυθμίζει ποίκιλες κυτταρικές λειτουργίες. Το PTEN δρώντας ως φωσφατάση αποφωσφορυλιώνει και μειώνει τα επίπεδα του PIP3 και αυξάνει τα επίπεδα του PIP2, οδηγώντας σε μείωση της δραστηριότητας της AKT. Πηγή: (94).
1.3. Hurthle ή oγκοκυτταρικός καρκίνος θυρεοειδούς
Αν και το νεόπλασμα αυτό θεωρούνταν παλιά ξεχωριστή κλινική οντότητα η πρόσφατη WHO/AIRC (Italian Association for the Research against Cancer) ταξινόμηση το κατατάσει σαν υποτύπο του θυλακιώδους καρκίνου θυρεοειδούς που χαρακτηρίζεται από συσσώρευση στο κυτταρόπλασμα μιτοχονδρίων με μορφολογικές διαταραχές (100). Σε σύγκριση με το τελευταίο είναι ανθεκτικό συνήθως στη θεραπεία με ιώδιο και σχετίζεται με μεγαλύτερη προδιάθεση για λεμφαδενικές μεταστάσεις.
Η κύρια μοριακή γενετική διαταραχή των όγκων Hurthle αφορά τις μιτοχονδριακές πρωτεΐνες που κωδικοποιούνται τόσο από μιτοχονδριακό DNA(101,102) όσο και από γονίδια του πυρήνα (103). Μεταλλάξεις του πυρηνικού γονιδίου GRIM-19 είναι ειδικές για όγκους Hurthle σποραδικούς ή οικογενείς, καθώς η πρωτεΐνη που κωδικοποιεί εμπλέκεται και στο μεταβολισμό των μιτοχονδρίων και στον κυτταρικό θάνατο (103).Υπάρχουν στοιχεία ότι η δυσλειτουργία της μιτοχονδριακής αλυσίδας (MRC) μπορεί να είναι επίκτητη κατά τη διαδικασία της μετατροπής των κυττάρων του θυρεοειδούς σε νεοπλασματικά. Για παράδειγμα η συνεχής ενεργοποίηση του ογκογονιδίου BRAF οδηγεί σε μειωμένη έκφραση ενός συνόλου ενζύμων του complex 1 της αναπνευστικής αλυσίδας των μιτοχονδρίων των κυττάρων του θυροειδούς(104).
1.4. Καρκίνος θυρεοειδούς χαμηλής διαφοροποίησης και αμετάπλαστο νεόπλασμα θυρεοειδούς
Το νεόπλασμα θυρεοειδούς χαμηλής διαφοροποίησης χαρακτηρίζεται από διηθητικό πρότυπο ανάπτυξης, νεκρώσεις, υψηλό μιτωτικό δείκτη και αγγειακή διήθηση (5). Το αναπλαστικό νεόπλασμα είναι υψηλής κακοήθειας όγκος και αποτελείται από αδιαφοροποίητα κύτταρα που διατηρούν τους δείκτες επιθηλιακής προέλευσης.
1.4.1. Μεταλλάξεις στο κυτταρικό μονοπάτι της ΜΑPK κινάσης
Στα νεοπλάσματα θυρεοειδούς φτωχής διαφοροποίησης μεταλλάξεις των ογκογονιδίων RAS ανευρίσκονται σε υψηλό ποσοστό (40-55%) (105). Αντίθετα μεταλλάξεις του ογκογονιδίου BRAF είναι λιγότερο συχνές (12-17%), ωστόσο επί υπάρξεως τους αυξάνει η θνητότητα των ασθενών (105).
Στο αμετάπλαστο νεόπλασμα του θυρεοειδούς οι κυρίαρχες μεταλλάξεις στο μονοπάτι της MAPK κινάσης αφορούν το ογκογονίδιο BRAF, με συχνότητα (25-50%) (105, 106). Αναδιατάξεις των RET/PTC, NTRK και PAX8–PPARγονιδίων σπάνια ανευρίσκονται στα χαμηλής διαφοροποίησης και στα αμετάπλασματα νεοπλάσματα θυρεοειδούς, υποστηρίζοντας την παρατήρηση ότι καλά διαφοροποιημένα νεοπλάσματα θυρεοειδούς που οφείλονται σε χιμαιρικά ογκογονίδια σπάνια εξελίσσονται σε όγκους υψηλής κακοήθειας (105).
1.4.2. Μεταλλάξεις στο κυτταρικό μονοπάτι PTEN/PI3K/Akt
Μεταλλάξεις στο μονοπάτι PTEN/PI3K/Akt ανευρίσκονται σε υψηλό ποσοστό στο αμετάπλαστο νεόπλασμα θυρεοειδούς (98). Ειδικότερα μεταλλάξεις στο γονίδιο που κωδικοποιεί την καταλυτική υπομονάδα της φωσφατιδυλινοσιτόλης-3 κινάσης(PI3K) ανευρίσκονται σε ποσοστό 23% και συχνά συνυπάρχουν με μεταλλάξεις των γονιδίων RAS ή BRAF (98). Μεταλλάξεις του ογκοκατασταλτικού γονιδίου PTEN που ενεργοποιούν το μονοπάτι PI3K/Akt ανευρίσκονται επίσης σε υψηλό ποσοστό (12-50%) σε αμετάπλαστο νεόπλασμα θυρεοειδούς (99).
Τέλος μεταλλάξεις της κινάσης σερίνης-θρεονίνης AKT1 ανευρίσκονται στο φτωχής διαφοροποίησης νεόπλασμα θυρεοειδούς, χωρίς να αλληλοκαλύπτονται με μεταλλάξεις της καταλυτικής υπομονάδας PI3K και φαίνεται ότι αποτελούν ένα εναλλακτικό μηχανισμό ενεργοποίησης του κυτταρικού αυτού μονοπατιού σε προχωρημένες μορφές καρκίνου θυρεοειδούς. Συχνά συνδέονται με μεταλλάξεις του γονιδίου BRAF (107).
1.4.3. Oγκοκατασταλτικό γονίδιο p53
Το γονίδιο TP53 κωδικοποιεί μία πρωτεΐνη του πυρήνα που προκαλεί αναστολή του κυτταρικού κύκλου, και/ή απόπτωση ως ανταπόκριση σε διάφορα ερεθίσματα. Απενεργοποίηση του μπορεί να οδηγήσει σε καρκινογένεση, εξέλιξη της νόσου και αντίσταση στην αντινεοπλασματική θεραπεία. Αν και μεταλλάξεις του p53 ανευρίσκονται σπάνια σε καλά διαφοροποιημένα νεοπλάσματα θυρεοειδούς (0-9%) είναι συχνές στα νεοπλάσματα χαμηλής διαφοροποίησης (17-38%) και ιδίως στα αμετάπλασματα νεοπλάσματα όπου το ποσοστό φτάνει στο 67-88% (108). Φαίνεται ότι αποτελεί ένα όψιμο γεγονός στη διαδικασία της καρκινογένεσης στον θυρεοειδή και ότι σχετίζεται με πιο επιθετικό φαινότυπο.
1.4.4. Μεταλλάξεις στη beta–catenin και στο κυτταρικό μονοπάτι
Η πρωτεΐνη β-catenin κωδικοποιείται από το γονίδιο CTNNB1 και παίζει ρόλο τόσο στην σύζευξη των κυττάρων όσο και στην μεταγραφή στον πυρήνα (109). Στο φυσιολογικό κύτταρο η β-catenin είναι κυρίως προσδεδεμένη στην κυτταρική μεμβράνη με τις διαμεμβρανικές πρωτεΐνες cadherins. Επίσης αποτελεί σημαντικό στοιχείο του Wnt μονοπατιού σηματοδότησης το οποίο είναι απαραίτητο για την ανάπτυξη του εμβρύου, αλλά ενεργοποιείται και σε πολλούς καρκίνους στον άνθρωπο (109).
Η b-catenin είναι τμήμα ενός κυτταροπλασματικού συμπλέγματος που επίσης περιλαμβάνει την APC (adenomatosis polyposis coli) και την αξίνη που απενεργοποιείται από την συνθάση του γλυκογόνου κινάση-3 (GSK3b). To Wnt μονοπάτι σταθεροποιεί την β-catenin εμποδίζοντας την φωσφορυλίωση της από την GSK-3 και την επακόλουθη αποδόμησή της και της επιτρέπει να μετακινηθεί στον πυρήνα όπου δρα ως συνενεργοποιητής των μεταγραφικών παραγόντων που ανήκουν στην οικογένεια TCF/LEF.
Μεταλλάξεις στο γονίδιο που κωδικοποιεί την APC είναι υπεύθυνες για την ανάπτυξη της οικογενούς αδενωματώδους πολυποδίασης και της ποικιλίας του το σύνδρομο Gardner που συνδυάζονται με αυξημένο κίνδυνο ανάπτυξης θηλώδους καρκινώματος θυρεοειδούς (110). Από την άλλη πλευρά η έκφραση της e-cadherin είναι αυξημένη στα φυσιολογικά κύτταρα του θυρεοειδούς, αλλά μειωμένη στο αμετάπλαστο καρκίνωμα (111). Επίσης μεταλλάξεις της β-catenin, που οδηγούν στην εναπόθεσή της στον πυρήνα και στο συνεχή ρόλο της ως μεταγραφικό συνενεργοποιητή ανευρίσκονται στα καρκινώματα θυρεοειδούς φτωχής διαφοροποίησης (25%) και στα αμετάπλαστα (65%), αλλά όχι στα καλά διαφοροποιημένα καρκινώματα (112,113)
2. Μοριακοί δείκτες βασισμένοι στο RNA
2.1. MicroRNA (miRNA)
Τα miRNA είναι τμήματα 21-22 νουκλεοτιδίων μη κωδικοποιημένου RNA που έχουν σημαντικό ρόλο κυρίως ανασταλτικό στη μετα-μεταγραφική ρύθμιση γονιδίων μέσω συμπληρωματικής πρόσδεσης στο mRNA, που μεσολαβεί στην μετάφραση και αποδόμηση του. Πολλές μελέτες έχουν βρει αυξημένη έκφραση διαφορετικών miRNA στο θηλώδες καρκίνωμα του θυρεοειδούς (κυρίως τα miR-221 και -222) (114,115).
Μέχρι τώρα δεν υπάρχει μεγάλη, προοπτική, πολυκεντρική μελέτη για τη χρησιμότητα των miRNA και συνήθως χρησιμοποιούνται μόνο σε ερευνητικά πρωτόκολλα.
2.2. Προσδιορισμός του mRNA του υποδοχέα της TSH (TSHR)
Tα καρκινικά κύτταρα του θυρεοειδούς μπορούν να εκφράζουν λειτουργικούς υποδοχείς της TSH (TSHR). Το mRNA του TSHR χρησιμοποιείται ως δείκτης στο περιφερικό αίμα στην παρακολούθηση ασθενών με καλά διαφοροποιημένο καρκίνο θυρεοειδούς.
Όσον αφορά την προεγχειρητική διάγνωση νεοπλασίας σε όζους θυρεοειδή μία μελέτη έδειξε ότι ο προσδιορισμός του TSHR mRNA έχει ευαισθησία 90% και ειδικότητα 80% για την πρόβλεψη κακοήθειας ειδικά σε ασθενείς με αδιευκρίνιστη κυτταρολογική εξέταση(intermediate) μετά FNA (116).
2.3. Ανάλυση της έκφρασης γονιδίων (micro array analysis)
Aντίθετα με τις τεχνικές ανίχνευσης μεταλλάξεων σε ένα γονίδιο ή αναδιατάξεων οι microarray διαγνωστικές δοκιμασίες μπορούν να ανιχνεύσουν την έκφραση εκατοντάδων γονιδίων από το mRNA που απομονώνεται από το ξέπλυμα της βελόνας κατά τη διάρκεια της FNA.
Mεγάλη προοπτική, πολυκεντρική μελέτη που μελέτησε την έκφραση 167 γονιδίων με την τεχνική αυτή έδειξε ότι είχε ευαισθησία 92% και ειδικότητα 52% στη διάγνωση κακοήθειας (117).
3. Ανοσοϊστοχημικοί δείκτες
Τα τελευταία χρόνια διάφοροι ανοσοιστοχημικοί δείκτες στον ορό των ασθενών όπως galectin-3, HBME-1, fibronectin-, CITED-1, και cytokeratin-19 έχουν διερευνηθεί για το ρόλο τους στη διάκριση μεταξύ καλοήθων και κακοήθων οζιδίων θυρεοειδούς. Ωστόσο δεν χρησιμοποιούνται ευρέως στην κλινική πράξη με εξαίρεση την galectin-3 καθώς δείχνουν σημαντική αλληλοεπικάλυψη μεταξύ της διάγνωσης θυλακιώδους αδενώματος και καλά διαφοροποιημένου καρκίνου θυρεοειδούς (118).
Galectin-3. H Galectin-3 ανήκει στη μεγάλη οικογένεια των Galectins πρωτεϊνών που αναγνωρίζουν και προσδένουν τις γαλακτοσίδες από τις κυτταρικές γλυκοπρωτείνες και γλυκολιπίδια (119). Εκφράζεται στον πυρήνα, στο κυτταρόπλασμα, στα μιτοχόνδρια, στην κυτταρική επιφάνεια και στο εξωκυττάριο διάστημα και φαίνεται να εμπλέκεται εκτός των άλλων στην αύξηση και διαφοροποίηση του κυττάρου, στον κυτταρικό κύκλο και στην απόπτωση (120). H Galectin-3 δεν εκφράζεται σε φυσιολογικά κύτταρα θυρεοειδούς αλλά είναι δυνατόν να εκφραστεί σε θυλακιώδη αδενώματα (0-45%), ενώ αποτελεί έναν από τους πιο αξιόπιστους μοριακούς δείκτες για το καλά διαφοροποιημένο νεόπλασμα θυρεοειδούς (76,121).
Στις περισσότερες μελέτες αναφέρεται έκφραση της Galectin-3 στο 90-100% θηλωδών νεοπλασμάτων θυρεοειδούς με την ανεύρεσή της να ποικίλλει ανάλογα με τον ιστολογικό υπότυπο. Στο κλασσικό θηλώδες νεόπλασμα ανευρίσκεται σε ποσοστό 82-100% ενώ στο θηλώδες με θυλακιώδη διαμόρφωση η έκφρασή της κυμαίνεται από 33% έως 100%.
Στα θυλακιώδη νεοπλάσματα η Galectin-3 εκφράζεται σε μικρότερο ποσοστό (20-100%) από ότι στα θηλώδη (121).
4. Ρόλος των μοριακών δεικτών στην πρόγνωση του καρκίνου του θυρεοειδούς
Οι μοριακοί δείκτες μπορούν να παίξουν σημαντικό ρόλο στην πρόγνωση της πορείας του καρκίνου του θυρεοειδούς. Είναι γνωστό ότι μεταλλάξεις των γονιδίων RAS, PIK3CA, and PTEN συχνά συμβαίνουν και συνυπάρχουν κατά την εξέλιξη ενός όγκου του θυρεοειδούς από αρχικό σε προχωρημένο στάδιο (63). Επίσης γενετικά patterns, ιδίως συνύπαρξη των ανωτέρω με άλλες γενετικές διαταραχές (π.χ. μεταλλάξεις του γονιδίου ΒRAF) που μπορούν να ενεργοποιήσουν και τα δύο σημαντικά μονοπάτια σηματοδότησης MAPK και PI3K-AKT, συχνά συμβαίνουν κατά την εξέλιξη καρκίνου του θυρεοειδούς σε προχωρημένο στάδιο (63,122). Αυτά τα ευρήματα είναι ενδεικτικά ότι τέτοια γενετικά patterns θα μπορούσαν να αποτελέσουν δείκτες φτωχής πρόγνωσης καρκίνου του θυρεοειδούς.
Μεταλλάξεις του ογκογονιδίου RAS, κυρίως του NRAS, συνδέονται με αυξημένη επιθετικότητα θυλακιώδους και φτωχής διαφοροποίησης νεοπλάσματος θυρεοειδούς και με μείωση της συνολικής επιβίωσης των ασθενών (123,124). Επίσης μεταλλάξεις συγκεκριμένων ογκογονιδίων όπως το p53 ανευρίσκονται μόνο στο φτωχής διαφοροποίησης ή στο αμετάπλαστο νεόπλασμα θυρεοειδούς, ενώ άλλες όπως το AKT1 έχουν αναφερθεί μόνο σε μεταστάσεις και όχι στις πρωτοπαθείς εστίες στο θυρεοειδή (107,102). Η προγνωστική χρησιμότητα των ανωτέρω γενετικών δεικτών αναμένεται να διευκρινιστεί.
Ο πιο καλά καθορισμένος προγνωστικός δείκτης για το θηλώδες νεόπλασμα θυρεοειδούς είναι μεταλλάξεις του ογκογονιδίου BRAF που ενεργοποιούν το μονοπάτι της ΜΑPK κινάσης. Μεγάλη πολυκεντρική μελέτη, αλλά και μεταγενέστερες μελέτες έδειξαν ισχυρή συσχέτιση μεταλλάξεων του BRAF με ανάπτυξη λεμφαδενικών μεταστάσεων, εξωθυρεοειδική επέκταση, προχωρημένα στάδια (ΙΙΙ, ΙV) καθώς και υποτροπή της νόσου (59,125) ακόμη και σε ασθενείς χαμηλού κινδύνου (59,126).
Επίσης, όπως έχει ήδη αναφερθεί μεταλλάξεις του ογκογονιδίου BRAF συνδέονται με θηλώδες νεόπλασμα θυρεοειδούς ανθεκτικό στο θεραπευτικό ιώδιο, καθώς η ύπαρξή τους συσχετίζεται με μείωση ή απώλεια της έκφρασης στον θυρεοειδικό ιστό γονιδίων που δεσμεύουν το ιώδιο όπως τα γονίδια για το NIS, TSHR, pendrin, TPO και TG (127).
Τέλος, μεταλλάξεις του ογκογονιδίου BRAF φαίνεται ότι προκαλούν υπερέκφραση άλλων ογκογονιδίων όπως των VEGF και MET (127). Τα ανωτέρω ευρήματα εξηγούν τη συσχέτιση της υψηλής επιθετικότητας νεοπλασμάτων θυρεοειδούς που φέρουν μεταλλάξεις του ογκογονιδίου BRAF καθώς και τη συσχέτισή τους με αυξημένη θνητότητα των ασθενών που τις έχουν, όπως έχει φανεί σε διεθνή πολυκεντρική μελέτη (128).
Γενετικές μεταλλάξεις και ο προγνωστικός τους ρόλος στον οικογενή καλά διαφοροποιημένο καρκίνο του θυρεοειδούς. Από το σύνολο των περιπτώσεων καρκίνου του θυρεοειδούς 5-10% εμφανίζουν γενετική προδιάθεση και κληρονομούνται με τον αυτοσωματικό κυρίαρχο τύπο (129). Το 85% των περιπτώσεων αποτελούν οικογενές θηλώδες νεόπλασμα θυρεοειδούς το οποίο διαφέρει από τις σποραδικές μορφές στο ότι εμφανίζεται σε νεαρότερη ηλικία, είναι πιο συχνά πολυεστιακό και έχει υψηλότερο βαθμό επιθετικότητας και υψηλότερη συχνότητα υποτροπής.
Η γενετική βάση του οικογενούς καρκίνου θυρεοειδούς είναι ετερογενής (130,131). Σωματικές μεταλλάξεις των γονιδίων BRAF ή RAS ανευρίσκονται στο οικογενές θηλώδες νεόπλασμα θυρεοειδούς σε ίδια συχνότητα με τις σποραδικές μορφές (129). Ωστόσο φαίνεται ότι η συχνότητα θηλωδών οικογενών νεοπλασμάτων συσχετιζόμενων με μετάλλαξη στο BRAF ογκογονίδιο αυξάνει από περιβαλλοντικούς παράγοντες, όπως η αυξημένη πρόσληψη ιωδίου (132).
5. Ρόλος των μοριακών δεικτών στην προεγχειρητική διάγνωση του καρκίνου του θυρεοειδούς
Ο κίνδυνος κακοήθειας σε αρνητικά κυτταρολογικά δείγματα μετά από παρακέντηση όζων θυρεοειδούς είναι μικρότερος από 3% επομένως η χρήση των μοριακών δεικτών σε αυτούς τους ασθενείς δεν είναι τόσο σημαντική. Αντίθετα στα κυτταρολογικά δείγματα με ατυπία (indeterminate) η ανάλυση για μετάλλαξη σε μοριακούς δείκτες μπορεί να είναι ιδιαίτερα χρήσιμη. Συγκεκριμένα ανεύρεση μετάλλαξης στο ογκογονίδιο BRAF είναι διαγνωστική για θηλώδες νεόπλασμα θυρεοειδούς 133. Το ίδιο ισχύει και με το χιμαιρικό ογκογονίδιο RET/PTC που είναι ειδικό για το θηλώδες νεόπλασμα. Αντίθετα ανεύρεση μεταλλάξεων του RAS δεν προσφέρουν σημαντικά καθώς μπορεί να είναι ενδεικτικές θηλώδους ή θυλακιώδους νεοπλάσματος θυρεοειδούς ή καλοήθους θυλακιώδους αδενώματος. Μελέτες έχουν δείξει ότι η ταυτόχρονη αναζήτηση μεταλλάξεων ενός συνόλου μοριακών δεικτών (BRAF, RET/PTC, Ras, και PAX8/PPARG1) σε δείγματα FNA που χαρακτηρίζονται ως θυλακιώδους μορφολογίας ή αδιευκρίνιστα, αν είναι θετική, μπορεί να λειτουργήσει συμπληρωματικά για την ορθή διάγνωση και αντιμετώπιση των ασθενών (134).
Μια εναλλακτική προσέγγιση των αδιευκρίνιστων δειγμάτων κυτταρολογικής εξέτασης περιλαμβάνει την προσθήκη στα ανωτέρω, ανάλυσης miRNA καθώς και προσδιορισμό βιοδεικτών στον ορό των ασθενών, περιλαμβανομένων και πρωτεϊνών που προέρχονται από τα ανωτέρω ογκογονίδια (135,136).
6. Ρόλος των μοριακών δεικτών στη θεραπεία του καρκίνου του θυρεοειδούς
Οι μοριακοί δείκτες μπορούν να παίξουν καθοριστικό ρόλο στις θεραπευτικές αποφάσεις για τον καρκίνο του θυρεοειδούς.
H προεγχειρητική ανεύρεση μετάλλαξης στο ογκογονίδιο BRAF μπορεί να βοηθήσει στην αναγνώριση μικροκαρκινωμάτων θυρεοειδούς που δυνητικά έχουν κακή πρόγνωση και συχνές υποτροπές και να οδηγήσει στην πιο επιθετική αντιμετώπισή τους. Συγκεκριμένα μπορεί να οδηγήσει στην απόφαση για χορήγηση ραδιενεργού ιωδίου για καταστροφή του υπολείμματος του θυρεοειδούς μετά θυρεοειδεκτομή (ablation) ακόμη και σε ασθενείς χαμηλού κινδύνου (126) αποκαθιστώντας τη θυρεοεσφαιρίνη (Tg) ως αξιόπιστο καρκινικό δείκτη παρακολούθησης. Επίσης ενδέχεται να βοηθήσει στην απόφαση για ταυτόχρονο κεντρικό λεμφαδενικό καθαρισμό μαζί με ολική θυρεοειδεκτομή, καθώς η ύπαρξη του σχετίζεται με υποτροπή θηλώδους καρκινώματος συνήθως σε λεμφαδένες του κεντρικού διαμερίσματος σε ποσοστό 75-95% και επανεγχείρηση στην περιοχή (137,138).
Ένας άλλος σημαντικός ρόλος των μοριακών δεικτών είναι η αντιμετώπιση ασθενών με μεταστατικό καρκίνο θυρεοειδούς ανθεκτικό στην θεραπεία με ραδιενεργό ιώδιο. Ο στόχος των κλινικών μελετών που διεξάγονται σήμερα είναι η ανεύρεση μίας γενετικής διαταραχής και η στοχευμένη φαρμακευτική θεραπεία στους ανωτέρω ασθενείς (139). Mέχρι τώρα μελετώνται ευρέως φάρμακα έναντι της αγγειογένεσης καθώς και αναστολείς των κινασών του μονοπατιού MAPK (Εικόνα 7). H αντινεοπλασματική ικανότητα αυτών των παραγόντων φαίνεται να είναι περισσότερο αποτελεσματική από τις κυτταροτοξικές θεραπείες με μερική ανταπόκριση σε 0-59% των ασθενών σε κλινικές μελέτες φάσης 2 και μακροχρόνια σταθεροποίηση της νόσου σε άλλο ένα τρίτο των ασθενών (140,141,142). Τα περισσότερα φάρμακα είναι πιο αποτελεσματικά σε μεταστάσεις που εντοπίζονται σε λεμφαδένες, ήπαρ, και πνεύμονες παρά στα οστά. Ωστόσο απαιτούνται περισσότερες μελέτες σε πειραματικά μοντέλα για να διερευνηθεί η συσχέτιση μεταξύ της αποτελεσματικότητας του φαρμάκου και της γενετική διαταραχής που ανευρίσκεται στον όγκο.
Ένας άλλος τρόπος αντιμετώπισης αυτών των ασθενών είναι η αποκατάσταση της ικανότητας πρόσληψης ιωδίου από τα καρκινικά κύτταρα και ακολούθως η χορήγηση ραδιενεργού ιωδίου μετά προετοιμασία με TSH. Αυτή η προσέγγιση υποστηρίχτηκε από in vitro μελέτες όπου η χορήγηση αναστολέων των γονιδίων BRAF, MEK, ή AKT στα μονοπάτια MAPK και PI3K-AKT μπορούσε να αποκαταστήσει την έκφραση γονιδίων στον θυρεοειδή και την πρόσληψη ραδιενεργού ιωδίου (143). Τα ευρήματα αυτά επιβεβαιώθηκαν αργότερα και με μελέτες in vivo (144,145). Tα κλινικά στοιχεία είναι ιδιαίτερα ενθαρρυντικά για τον αναστολέα της ΜΕΚ κινάσης selumetinib ιδιαίτερα σε καρκίνους θυρεοειδή που φέρουν μεταλλάξεις του ογκογονιδίου RAS (145).
Οι ανεπιθύμητες ενέργειες των αναστολέων των κινασών (κακουχία, διάρροια, υπέρταση και δερματικά εξανθήματα) έχουν οδηγήσει σε μείωση της δόσης στο 11-73% των ασθενών και σε διακοπή του φαρμάκου στο 7-25% των ασθενών. Επομένως θα πρέπει να χορηγούνται σε επιλεγμένους ασθενείς με σημαντικό φορτίο όγκου και εκτεταμένη νόσο όπου η πιθανότητα κλινικής ανταπόκρισης σε συγκεκριμένη θεραπεία να είναι υψηλή με βάση την ανεύρεση μεταλλάξεων στους ανωτέρω μοριακούς δείκτες.
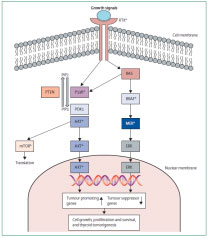
Εικόνα 7. MAPK και PI3K-AKT-MTOR μονοπάτια – γενετικές μεταλλάξεις και θεραπευτικοί στόχοι στον καρκίνο του θυρεοειδούς. Τα δύο κλασσικά μονοπάτια σηματοδότησης είναι σε σύζευξη με τον υποδοχέα τυροσινικής κινάσης (RTK) στην επιφάνεια του κυττάρου ο οποίος μεταφέρει τα εξωκυττάρια μηνύματα αύξησης σε ενδοκυττάρια μηνύματα διαμέσου των μονοπατιών αυτών. *Υποδεικνύει τους θεραπευτικούς στόχους που εξετάζονται σήμερα κλινικά στον καρκίνο του θυρεοειδούς. Πηγή: (146).
Βιβλιογραφία
1. Shortt J, Johnstone RW. Oncogenes in cell survival and cell death. Cold Spring Harb Perspect Biol 2012;4(12).
2. Balta AZ, Filiz AI, Kurt Y, Sucullu I, Yucel E, Akin ML. Prognostic value of oncoprotein expressions in thyroid papillary carcinoma. Med Oncol 2012;29(2):734-41.
3. Krohn K, Fuhrer D, Bayer Y, Eszlinger M, Brauer V, Neumann S, et al. Molecular pathogenesis of euthyroid and toxic multinodular goiter. Endocr Rev 2005;26(4):504-24.
4. Sastre-Perona A, Santisteban P. Role of the wnt pathway in thyroid cancer. Front Endocrinol (Lausanne) 2012;3:31.
5. DeLellis RA LR, Heitz PU, Eng C, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. Lyon: IARC, 2004.
6. Nikiforov YE. Thyroid carcinoma: molecular pathways and therapeutic targets. Mod Pathol 2008;21 Suppl 2:S37-43.
7. Gandhi M, Evdokimova V, Nikiforov YE. Mechanisms of chromosomal rearrangements in solid tumors: the model of papillary thyroid carcinoma. Mol Cell Endocrinol 2010;321(1):36-43.
8. Bhaijee F, Nikiforov YE. Molecular analysis of thyroid tumors. Endocr Pathol 2011;22(3):126-33.
9. Nikiforov YE. Molecular diagnostics of thyroid tumors. Arch Pathol Lab Med 2011;135(5):569-77.
10. Handkiewicz-Junak D, Czarniecka A, Jarzab B. Molecular prognostic markers in papillary and follicular thyroid cancer: Current status and future directions. Mol Cell Endocrinol 2010;322(1-2):8-28.
11. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer 2005;12(2):245-62.
12. Romei C, Elisei R. RET/PTC Translocations and Clinico-Pathological Features in Human Papillary Thyroid Carcinoma. Front Endocrinol (Lausanne) 2012;3:54.
13. Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev 2005;16(4-5):441-67.
14. Knowles PP, Murray-Rust J, Kjaer S, Scott RP, Hanrahan S, Santoro M, et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem 2006;281(44):33577-87.
15. Baloh RH, Enomoto H, Johnson EM, Jr., Milbrandt J. The GDNF family ligands and receptors – implications for neural development. Curr Opin Neurobiol 2000;10(1):103-10.
16. Santoro M, Rosati R, Grieco M, Berlingieri MT, D’Amato GL, de Franciscis V, et al. The ret proto-oncogene is consistently expressed in human pheochromocytomas and thyroid medullary carcinomas. Oncogene 1990;5(10):1595-8.
17. Fusco A, Grieco M, Santoro M, Berlingieri MT, Pilotti S, Pierotti MA, et al. A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature 1987;328(6126):170-2.
18. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 1990;60(4):557-63.
19. Caudill CM, Zhu Z, Ciampi R, Stringer JR, Nikiforov YE. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: a model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab 2005;90(4):2364-9.
20. Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol 1994;65(1):7-17.
21. Gandhi M, Dillon LW, Pramanik S, Nikiforov YE, Wang YH. DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene 2010;29(15):2272-80.
22. Narayanan PK, Goodwin EH, Lehnert BE. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res 1997;57(18):3963-71.
23. Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol 2002;13(1):3-16.
24. Soares P, Fonseca E, Wynford-Thomas D, Sobrinho-Simoes M. Sporadic ret-rearranged papillary carcinoma of the thyroid: a subset of slow growing, less aggressive thyroid neoplasms? J Pathol 1998;185(1):71-8.
25. Viglietto G, Chiappetta G, Martinez-Tello FJ, Fukunaga FH, Tallini G, Rigopoulou D, et al. RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene 1995;11(6):1207-10.
26. Hamatani K, Eguchi H, Ito R, Mukai M, Takahashi K, Taga M, et al. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res 2008;68(17):7176-82.
27. Hieber L, Huber R, Bauer V, Schaffner Q, Braselmann H, Thomas G, et al. Chromosomal rearrangements in post-Chernobyl papillary thyroid carcinomas: evaluation by spectral karyotyping and automated interphase FISH. J Biomed Biotechnol 2011;2011:693691.
28. Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res 1997;57(9):1690-4.
29. Fenton CL, Lukes Y, Nicholson D, Dinauer CA, Francis GL, Tuttle RM. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J Clin Endocrinol Metab 2000;85(3):1170-5.
30. Zhu Z, Ciampi R, Nikiforova MN, Gandhi M, Nikiforov YE. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: effects of the detection methods and genetic heterogeneity. J Clin Endocrinol Metab 2006;91(9):3603-10.
31. Unger K, Zitzelsberger H, Salvatore G, Santoro M, Bogdanova T, Braselmann H, et al. Heterogeneity in the distribution of RET/PTC rearrangements within individual post-Chernobyl papillary thyroid carcinomas. J Clin Endocrinol Metab 2004;89(9):4272-9.
32. Sapio MR, Guerra A, Marotta V, Campanile E, Formisano R, Deandrea M, et al. High growth rate of benign thyroid nodules bearing RET/PTC rearrangements. J Clin Endocrinol Metab 2011;96(6):E916-9.
33. Rhoden KJ, Unger K, Salvatore G, Yilmaz Y, Vovk V, Chiappetta G, et al. RET/papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: follicular cells of Hashimoto’s thyroiditis share low-level recombination events with a subset of papillary carcinoma. J Clin Endocrinol Metab 2006;91(6):2414-23.
34. Djakiew D, Delsite R, Pflug B, Wrathall J, Lynch JH, Onoda M. Regulation of growth by a nerve growth factor-like protein which modulates paracrine interactions between a neoplastic epithelial cell line and stromal cells of the human prostate. Cancer Res 1991;51(12):3304-10.
35. Otten U, Ehrhard P, Peck R. Nerve growth factor induces growth and differentiation of human B lymphocytes. Proc Natl Acad Sci U S A 1989;86(24):10059-63.
36. Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 1986;319(6056):743-8.
37. Bounacer A, Schlumberger M, Wicker R, Du-Villard JA, Caillou B, Sarasin A, et al. Search for NTRK1 proto-oncogene rearrangements in human thyroid tumours originated after therapeutic radiation. Br J Cancer 2000;82(2):308-14.
38. Rabes HM, Demidchik EP, Sidorow JD, Lengfelder E, Beimfohr C, Hoelzel D, et al. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clin Cancer Res 2000;6(3):1093-103.
39. Bongarzone I, Vigneri P, Mariani L, Collini P, Pilotti S, Pierotti MA. RET/NTRK1 rearrangements in thyroid gland tumors of the papillary carcinoma family: correlation with clinicopathological features. Clin Cancer Res 1998;4(1):223-8.
40. Brzezianska E, Karbownik M, Migdalska-Sek M, Pastuszak-Lewandoska D, Wloch J, Lewinski A. Molecular analysis of the RET and NTRK1 gene rearrangements in papillary thyroid carcinoma in the Polish population. Mutat Res 2006;599(1-2):26-35.
41. Fedele M, Palmieri D, Chiappetta G, Pasquinelli R, De Martino I, Arra C, et al. Impairment of the p27kip1 function enhances thyroid carcinogenesis in TRK-T1 transgenic mice. Endocr Relat Cancer 2009;16(2):483-90.
42. Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 2003;4(5):373-84.
43. Nikiforova MN, Nikiforov YE. Molecular diagnostics and predictors in thyroid cancer. Thyroid 2009;19(12):1351-61.
44. Ezzat S, Zheng L, Kolenda J, Safarian A, Freeman JL, Asa SL. Prevalence of activating ras mutations in morphologically characterized thyroid nodules. Thyroid 1996;6(5):409-16.
45. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003;3(1):11-22.
46. Namba H, Rubin SA, Fagin JA. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol Endocrinol 1990;4(10):1474-9.
47. Vasko VV, Gaudart J, Allasia C, Savchenko V, Di Cristofaro J, Saji M, et al. Thyroid follicular adenomas may display features of follicular carcinoma and follicular variant of papillary carcinoma. Eur J Endocrinol 2004;151(6):779-86.
48. Lee SR, Jung CK, Kim TE, Bae JS, Jung SL, Choi YJ, et al. Molecular Genotyping of Follicular Variant of Papillary Thyroid Carcinoma Correlates with Diagnostic Category of Fine-Needle Aspiration Cytology: Values of RAS Mutation Testing. Thyroid 2013.
49. Howitt BE, Jia Y, Sholl LM, Barletta JA. Molecular alterations in partially-encapsulated or well-circumscribed follicular variant of papillary thyroid carcinoma. Thyroid 2013;23(10):1256-62.
50. Hara H, Fulton N, Yashiro T, Ito K, DeGroot LJ, Kaplan EL. N-ras mutation: an independent prognostic factor for aggressiveness of papillary thyroid carcinoma. Surgery 1994;116(6):1010-6.
51. Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, et al. The cardiofaciocutaneous syndrome. J Med Genet 2006;43(11):833-42.
52. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417(6892):949-54.
53. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116(6):855-67.
54. Adeniran AJ, Zhu Z, Gandhi M, Steward DL, Fidler JP, Giordano TJ, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol 2006;30(2):216-22.
55. Rivera M, Ricarte-Filho J, Knauf J, Shaha A, Tuttle M, Fagin JA, et al. Molecular genotyping of papillary thyroid carcinoma follicular variant according to its histological subtypes (encapsulated vs infiltrative) reveals distinct BRAF and RAS mutation patterns. Mod Pathol 2010;23(9):1191-200.
56. Nikiforova MN, Kimura ET, Gandhi M, Biddinger PW, Knauf JA, Basolo F, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab 2003;88(11):5399-404.
57. Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003;22(29):4578-80.
58. Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest 2005;115(1):94-101.
59. Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab 2005;90(12):6373-9.
60. Kim TY, Kim WB, Rhee YS, Song JY, Kim JM, Gong G, et al. The BRAF mutation is useful for prediction of clinical recurrence in low-risk patients with conventional papillary thyroid carcinoma. Clin Endocrinol (Oxf) 2006;65(3):364-8.
61. Riesco-Eizaguirre G, Gutierrez-Martinez P, Garcia-Cabezas MA, Nistal M, Santisteban P. The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I- targeting to the membrane. Endocr Relat Cancer 2006;13(1):257-69.
62. Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, et al. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res 2005;65(10):4238-45.
63. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer 2013;13(3):184-99.
64. Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991;251(4995):802-4.
65. Birchmeier C, Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol 1998;8(10):404-10.
66. Gentile A, Trusolino L, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev 2008;27(1):85-94.
67. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 1995;80(2):179-85.
68. Graziani A, Gramaglia D, Cantley LC, Comoglio PM. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem 1991;266(33):22087-90.
69. Boccaccio C, Ando M, Tamagnone L, Bardelli A, Michieli P, Battistini C, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998;391(6664):285-8.
70. Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, et al. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res 2002;62(7):2064-71.
71. Gude NA, Emmanuel G, Wu W, Cottage CT, Fischer K, Quijada P, et al. Activation of Notch-mediated protective signaling in the myocardium. Circ Res 2008;102(9):1025-35.
72. Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, et al. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol 1992;119(3):629-41.
73. Nardone HC, Ziober AF, LiVolsi VA, Mandel SJ, Baloch ZW, Weber RS, et al. c-Met expression in tall cell variant papillary carcinoma of the thyroid. Cancer 2003;98(7):1386-93.
74. Wiseman SM, Griffith OL, Melck A, Masoudi H, Gown A, Nabi IR, et al. Evaluation of type 1 growth factor receptor family expression in benign and malignant thyroid lesions. Am J Surg 2008;195(5):667-73; discussion 73.
75. Masago K, Asato R, Fujita S, Hirano S, Tamura Y, Kanda T, et al. Epidermal growth factor receptor gene mutations in papillary thyroid carcinoma. Int J Cancer 2009;124(11):2744-9.
76. Wiseman SM, Melck A, Masoudi H, Ghaidi F, Goldstein L, Gown A, et al. Molecular phenotyping of thyroid tumors identifies a marker panel for differentiated thyroid cancer diagnosis. Ann Surg Oncol 2008;15(10):2811-26.
77. Jo YS, Li S, Song JH, Kwon KH, Lee JC, Rha SY, et al. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J Clin Endocrinol Metab 2006;91(9):3667-70.
78. Tian X, Cong M, Zhou W, Zhu J, Liu Q. Relationship between protein expression of VEGF-C, MMP-2 and lymph node metastasis in papillary thyroid cancer. J Int Med Res 2008;36(4):699-703.
79. Di Palma T, Filippone MG, Pierantoni GM, Fusco A, Soddu S, Zannini M. Pax8 has a critical role in epithelial cell survival and proliferation. Cell Death Dis 2013;4:e729.
80. Wood WM, Sharma V, Bauerle KT, Pike LA, Zhou Q, Fretwell DL, et al. PPARgamma Promotes Growth and Invasion of Thyroid Cancer Cells. PPAR Res 2011;2011:171765.
81. Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, 2nd, Tallini G, et al. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab 2003;88(5):2318-26.
82. French CA, Alexander EK, Cibas ES, Nose V, Laguette J, Faquin W, et al. Genetic and biological subgroups of low-stage follicular thyroid cancer. Am J Pathol 2003;162(4):1053-60.
83. Dwight T, Thoppe SR, Foukakis T, Lui WO, Wallin G, Hoog A, et al. Involvement of the PAX8/peroxisome proliferator-activated receptor gamma rearrangement in follicular thyroid tumors. J Clin Endocrinol Metab 2003;88(9):4440-5.
84. Castro P, Rebocho AP, Soares RJ, Magalhaes J, Roque L, Trovisco V, et al. PAX8-PPARgamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab 2006;91(1):213-20.
85. Caria P, Dettori T, Frau DV, Di Oto E, Morandi L, Parmeggiani A, et al. Simultaneous occurrence of PAX8-PPARg and RET-PTC3 rearrangements in a follicular variant of papillary thyroid carcinoma. Am J Surg Pathol 2012;36(9):1415-20.
86. Reddi HV, McIver B, Grebe SK, Eberhardt NL. The paired box-8/peroxisome proliferator-activated receptor-gamma oncogene in thyroid tumorigenesis. Endocrinology 2007;148(3):932-5.
87. Nikiforov YE. Molecular analysis of thyroid tumors. Mod Pathol 2011;24 Suppl 2:S34-43.
88. Lemoine NR, Mayall ES, Wyllie FS, Williams ED, Goyns M, Stringer B, et al. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene 1989;4(2):159-64.
89. Vasko V, Ferrand M, Di Cristofaro J, Carayon P, Henry JF, de Micco C. Specific pattern of RAS oncogene mutations in follicular thyroid tumors. J Clin Endocrinol Metab 2003;88(6):2745-52.
90. Basolo F, Pisaturo F, Pollina LE, Fontanini G, Elisei R, Molinaro E, et al. N-ras mutation in poorly differentiated thyroid carcinomas: correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid 2000;10(1):19-23.
91. Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, et al. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol 2003;21(17):3226-35.
92. Schark C, Fulton N, Jacoby RF, Westbrook CA, Straus FH, 2nd, Kaplan EL. N-ras 61 oncogene mutations in Hurthle cell tumors. Surgery 1990;108(6):994-9; discussion 99-1000.
93. Esapa CT, Johnson SJ, Kendall-Taylor P, Lennard TW, Harris PE. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol (Oxf) 1999;50(4):529-35.
94. Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci 2008;121(Pt 3):249-53.
95. Fistarol SK, Anliker MD, Itin PH. Cowden disease or multiple hamartoma syndrome–cutaneous clue to internal malignancy. Eur J Dermatol 2002;12(5):411-21.
96. Harach HR, Soubeyran I, Brown A, Bonneau D, Longy M. Thyroid pathologic findings in patients with Cowden disease. Ann Diagn Pathol 1999;3(6):331-40.
97. Paes JE, Ringel MD. Dysregulation of the phosphatidylinositol 3-kinase pathway in thyroid neoplasia. Endocrinol Metab Clin North Am 2008;37(2):375-87, viii-ix.
98. Garcia-Rostan G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R, Hermsem MJ, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res 2005;65(22):10199-207.
99. Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr Relat Cancer 2009;16(1):17-44.
100. Maximo V, Sobrinho-Simoes M. Hurthle cell tumours of the thyroid. A review with emphasis on mitochondrial abnormalities with clinical relevance. Virchows Arch 2000;437(2):107-15.
101. Maximo V, Soares P, Lima J, Cameselle-Teijeiro J, Sobrinho-Simoes M. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: a study with emphasis on Hurthle cell tumors. Am J Pathol 2002;160(5):1857-65.
102. Gasparre G, Porcelli AM, Bonora E, Pennisi LF, Toller M, Iommarini L, et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci U S A 2007;104(21):9001-6.
103. Maximo V, Botelho T, Capela J, Soares P, Lima J, Taveira A, et al. Somatic and germline mutation in GRIM-19, a dual function gene involved in mitochondrial metabolism and cell death, is linked to mitochondrion-rich (Hurthle cell) tumours of the thyroid. Br J Cancer 2005;92(10):1892-8.
104. Mesa C, Jr., Mirza M, Mitsutake N, Sartor M, Medvedovic M, Tomlinson C, et al. Conditional activation of RET/PTC3 and BRAFV600E in thyroid cells is associated with gene expression profiles that predict a preferential role of BRAF in extracellular matrix remodeling. Cancer Res 2006;66(13):6521-9.
105. Costa AM, Herrero A, Fresno MF, Heymann J, Alvarez JA, Cameselle-Teijeiro J, et al. BRAF mutation associated with other genetic events identifies a subset of aggressive papillary thyroid carcinoma. Clin Endocrinol (Oxf) 2008;68(4):618-34.
106. Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab 2008;93(1):278-84.
107. Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res 2009;69(11):4885-93.
108. Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J Clin Invest 1993;91(4):1753-60.
109. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009;17(1):9-26.
110. Cetta F, Montalto G, Gori M, Curia MC, Cama A, Olschwang S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: results from a European cooperative study. J Clin Endocrinol Metab 2000;85(1):286-92.
111. von Wasielewski R, Rhein A, Werner M, Scheumann GF, Dralle H, Potter E, et al. Immunohistochemical detection of E-cadherin in differentiated thyroid carcinomas correlates with clinical outcome. Cancer Res 1997;57(12):2501-7.
112. Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G. Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol 2001;158(3):987-96.
113. Nikiforov YE. Genetic alterations involved in the transition from well-differentiated to poorly differentiated and anaplastic thyroid carcinomas. Endocr Pathol 2004;15(4):319-27.
114. Pallante P, Visone R, Croce CM, Fusco A. Deregulation of microRNA expression in follicular-cell-derived human thyroid carcinomas. Endocr Relat Cancer 2010;17(1):F91-104.
115. Nikiforova MN, Tseng GC, Steward D, Diorio D, Nikiforov YE. MicroRNA expression profiling of thyroid tumors: biological significance and diagnostic utility. J Clin Endocrinol Metab 2008;93(5):1600-8.
116. Chia SY, Milas M, Reddy SK, Siperstein A, Skugor M, Brainard J, et al. Thyroid-stimulating hormone receptor messenger ribonucleic acid measurement in blood as a marker for circulating thyroid cancer cells and its role in the preoperative diagnosis of thyroid cancer. J Clin Endocrinol Metab 2007;92(2):468-75.
117. Alexander EK, Kennedy GC, Baloch ZW, Cibas ES, Chudova D, Diggans J, et al. Preoperative diagnosis of benign thyroid nodules with indeterminate cytology. N Engl J Med 2012;367(8):705-15.
118. Ferraz C, Eszlinger M, Paschke R. Current state and future perspective of molecular diagnosis of fine-needle aspiration biopsy of thyroid nodules. J Clin Endocrinol Metab 2011;96(7):2016-26.
119. Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell 1994;76(4):597-8.
120. Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochim Biophys Acta 2006;1760(4):616-35.
121. Chiu CG, Strugnell SS, Griffith OL, Jones SJ, Gown AM, Walker B, et al. Diagnostic utility of galectin-3 in thyroid cancer. Am J Pathol 2010;176(5):2067-81.
122. Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab 2008;93(8):3106-16.
123. Fukahori M, Yoshida A, Hayashi H, Yoshihara M, Matsukuma S, Sakuma Y, et al. The associations between RAS mutations and clinical characteristics in follicular thyroid tumors: new insights from a single center and a large patient cohort. Thyroid 2012;22(7):683-9.
124. Volante M, Rapa I, Gandhi M, Bussolati G, Giachino D, Papotti M, et al. RAS mutations are the predominant molecular alteration in poorly differentiated thyroid carcinomas and bear prognostic impact. J Clin Endocrinol Metab 2009;94(12):4735-41.
125. Kim TH, Park YJ, Lim JA, Ahn HY, Lee EK, Lee YJ, et al. The association of the BRAF(V600E) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: a meta-analysis. Cancer 2012;118(7):1764-73.
126. Elisei R, Viola D, Torregrossa L, Giannini R, Romei C, Ugolini C, et al. The BRAF(V600E) mutation is an independent, poor prognostic factor for the outcome of patients with low-risk intrathyroid papillary thyroid carcinoma: single-institution results from a large cohort study. J Clin Endocrinol Metab 2012;97(12):4390-8.
127. Xing M. Prognostic utility of BRAF mutation in papillary thyroid cancer. Mol Cell Endocrinol 2010;321(1):86-93.
128. Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 2013;309(14):1493-501.
129. Cavaco BM, Batista PF, Martins C, Banito A, do Rosario F, Limbert E, et al. Familial non-medullary thyroid carcinoma (FNMTC): analysis of fPTC/PRN, NMTC1, MNG1 and TCO susceptibility loci and identification of somatic BRAF and RAS mutations. Endocr Relat Cancer 2008;15(1):207-15.
130. Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Sigurdsson A, Bergthorsson JT, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat Genet 2009;41(4):460-4.
131. Ngan ES, Lang BH, Liu T, Shum CK, So MT, Lau DK, et al. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J Natl Cancer Inst 2009;101(3):162-75.
132. Guan H, Ji M, Bao R, Yu H, Wang Y, Hou P, et al. Association of high iodine intake with the T1799A BRAF mutation in papillary thyroid cancer. J Clin Endocrinol Metab 2009;94(5):1612-7.
133. Cohen Y, Rosenbaum E, Clark DP, Zeiger MA, Umbricht CB, Tufano RP, et al. Mutational analysis of BRAF in fine needle aspiration biopsies of the thyroid: a potential application for the preoperative assessment of thyroid nodules. Clin Cancer Res 2004;10(8):2761-5.
134. Ohori NP, Nikiforova MN, Schoedel KE, LeBeau SO, Hodak SP, Seethala RR, et al. Contribution of molecular testing to thyroid fine-needle aspiration cytology of “follicular lesion of undetermined significance/atypia of undetermined significance”. Cancer Cytopathol 2010;118(1):17-23.
135. Chudova D, Wilde JI, Wang ET, Wang H, Rabbee N, Egidio CM, et al. Molecular classification of thyroid nodules using high-dimensionality genomic data. J Clin Endocrinol Metab 2010;95(12):5296-304.
136. Grogan RH, Mitmaker EJ, Clark OH. The evolution of biomarkers in thyroid cancer-from mass screening to a personalized biosignature. Cancers (Basel) 2010;2(2):885-912.
137. Tufano RP, Bishop J, Wu G. Reoperative central compartment dissection for patients with recurrent/persistent papillary thyroid cancer: efficacy, safety, and the association of the BRAF mutation. Laryngoscope 2012;122(7):1634-40.
138. Henderson YC, Shellenberger TD, Williams MD, El-Naggar AK, Fredrick MJ, Cieply KM, et al. High rate of BRAF and RET/PTC dual mutations associated with recurrent papillary thyroid carcinoma. Clin Cancer Res 2009;15(2):485-91.
139. Fagin JA. How thyroid tumors start and why it matters: kinase mutants as targets for solid cancer pharmacotherapy. J Endocrinol 2004;183(2):249-56.
140. Carr LL, Mankoff DA, Goulart BH, Eaton KD, Capell PT, Kell EM, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res 2010;16(21):5260-8.
141. Ahmed M, Barbachano Y, Riddell A, Hickey J, Newbold KL, Viros A, et al. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: a phase II study in a UK based population. Eur J Endocrinol 2011;165(2):315-22.
142. Leboulleux S, Bastholt L, Krause T, de la Fouchardiere C, Tennvall J, Awada A, et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol 2012;13(9):897-905.
143. Hou P, Bojdani E, Xing M. Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J Clin Endocrinol Metab 2010;95(2):820-8.
144. Chakravarty D, Santos E, Ryder M, Knauf JA, Liao XH, West BL, et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest 2011;121(12):4700-11.
145. Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med 2013;368(7):623-32.
146. Xing M, Haugen BR, Schlumberger M. Progress in molecular-based management of differentiated thyroid cancer. Lancet 2013;381(9871):1058-69.
Created: October 27, 2014
Last update: October 27, 2014