
Λήδα Σαρίκα
Βιολόγος
Ενδοκρινολογική Μονάδα Θεραπευτικής Κλινικής Πανεπιστημίου Αθηνών
Μαρία Αλεβιζάκη
Ενδοκρινολόγος
Ενδοκρινολογική Μονάδα Θεραπευτικής Κλινικής Πανεπιστημίου Αθηνών
1. Εισαγωγή
Το μυελοειδές καρκίνωμα του θυρεοειδούς (ΜΚΘ) είναι ένας σπάνιος κακοήθης όγκος ο οποίος περιγράφηκε για πρώτη φορά το 1959 από τον Hazard (Hazard et al 1959). Αποτελεί το 4-10% των νεοπλασμάτων του θυρεοειδούς, προέρχεται από τα παραθυλακιώδη C κύτταρα του αδένα των οποίων η εμβρυϊκή προέλευση είναι από τη νευρική ακρολοφία (crest). Το ΜΚΘ παράγει καλσιτονίνη η οποία αποτελεί και καρκινικό δείκτη.
Το 25-30% των ΜΚΘ παρουσιάζουν οικογενή επίπτωση και εμφανίζονται με κληρονομούμενη μορφή ενώ το υπόλοιπο 70-75% είναι σποραδικά.
2. Σύνδρομα Πολλαπλής Ενδοκρινολογικής Νεοπλασίας ΜΕΝ2
Η εμφάνιση της οικογενούς μορφής ΜΚΘ συνοδεύεται συχνά και από άλλες παθολογικές εκδηλώσεις κυρίως ενδοκρινών αδένων σαν συστατικό του Συνδρόμου Πολλαπλής Ενδοκρινικής νεοπλασίας ΜΕΝ2. Ανάλογα με τα συνοδά νοσήματα κατατάσσεται σε τρείς τύπους:
- Το σ. ΜΕΝ2Α όπου το ΜΚΘ είναι παρόν στο >90% των ασθενών και συνοδεύεται από φαιοχρωμοκύττωμα στο 40-50% των περιπτώσεων και υπερπαραθυρεοειδισμό στο 10-20% των ασθενών σπανιότερα δε από δερματική λειχηνοειδή αμυλοείδωση.
- Το σ. ΜΕΝ2Β στο οποίο το σύνολο σχεδόν των ασθενών (~100%) παρουσιάζει ΜΚΘ και συνοδεύεται από φαιοχρωμοκύττωμα στο 25% των περιπτώσεων και χαρακτηριστικά υποβλεννογόνια νευρώματα, εντερικά γαγγλιονευρώματα, σκελετικές δυσπλασίες και μαρφανοειδή εμφάνιση.
- Τα Οικογενή ΜΚΘ στα οποία η μόνη εκδήλωση είναι το ΜΚΘ. Τα οικογενή ΜΚΘ θεωρούνται σήμερα περισσότερο φαινοτυπικές παραλλαγές του συνδρόμου ΜΕΝ2Α με χαμηλή διεισδυτικότητα του φαιοχρωμοκυττώματος παρά μια διακριτή οντότητα (Kloos et al 2009).
3. Ογκογονίδιο RET
3.1. Εισαγωγικά στοιχεία
Η κληρονομούμενη μορφή του ΜΚΘ συνδέεται με μεταλλάξεις στο πρωτο-ογκογονίδιο RET. Το RET κωδικοποιεί μια διαμεμβρανική κινάση της τυροσίνης (TK) η οποία εμπλέκεται στην ανάπτυξη και διαφοροποίηση των ιστών που προέρχονται από την νευρική ακρολοφία κατά την εμβρυογένεση.
Το γονίδιο εδρεύει στο χρωμόσωμα 10, περιέχει 21 εξώνια σε μια περιοχή 60kb. Η πρωτεΐνη ret έχει τρείς περιοχές μία μεγάλη ενδοκυττάριο με δύο υποομάδες TK μία διαμεμβρανική περιοχή και μια εξωκυττάριο με τέσσερες περιοχές τύπου καδερίνης (cadherin) και μία πλούσια σε κυστεΐνες η οποία βρίσκεται κοντά στη μεμβράνη (Εικόνα 1).
Το γονίδιο RET ταυτοποιείται πρώτη φορά το 1985. Το 1993 συνδέονται αιτιολογικά μεταλλάξεις του με το σύνδρομο ΜΕΝ 2Α ενώ το 1994 συνδέεται με το σύνδρομο ΜΕΝ 2Β. Το πρώτο διεθνές consortium για την ανάλυση των μεταλλάξεων του γονιδίου RET γίνεται το 1996 στο Gubbio Ιταλίας (Eng et al 1996).
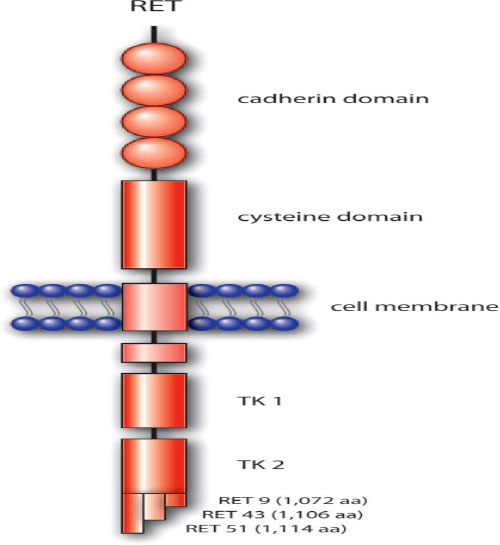
Εικόνα 1. Σχηματική παράσταση της πρωτεΐνης RET (De Groot et al 2006).
3.2. Ρόλος Ογκογονιδίου RET
Πιθανός φυσιολογικός του ρόλος είναι η διαβίβαση μηνυμάτων σε κύτταρα νευρικής προέλευσης κατά την ανάπτυξη, ρυθμίζει τον πολλαπλασιασμό, την μετανάστευση, την διαφοροποίηση και την επιβίωση των κυττάρων κατά τη διάρκεια της εμβρυογένεσης.
Έχουν περιγραφεί μεταλλάξεις του γονιδίου αυτού και στα κύτταρα του όγκου (σωματικές μεταλλάξεις) σε σποραδικά μυελοειδή καρκινώματα ενώ φαίνεται να μετέχει και στην παθογένεια του θηλώδους καρκινώματος του θυρεοειδούς στο οποίο υπάρχουν συχνά χρωμοσωμικές αναδιατάξεις στις οποίες συμμετέχει το γονίδιο RET (RET/PTC).
3.3. Η μεταβίβαση της νόσου στα σύνδρομα ΜΕΝ2
Στις οικογενείς περιπτώσεις του ΜΚΘ η μεταβίβαση της νόσου γίνεται με επικρατούντα αυτοσωμικό χαρακτήρα (κάθε απόγονος έχει 50% πιθανότητα να νοσήσει) και η μετάλλαξη είναι παρούσα σε όλα τα κύτταρα του οργανισμού σε ετερόζυγη μορφή στο σύνολο σχεδόν των περιπτώσεων. Η κάθε οικογένεια χαρακτηρίζεται από μια συγκεκριμένη γι αυτήν μετάλλαξη. Έχουν εντοπιστεί και αρκετοί πολυμορφισμοί του RET που έχουν συνδεθεί με την πορεία και την επιθετικότητα της νόσου.
3.4. Μεταλλάξεις του γονιδίου RET
Το 1993 για πρώτη φορά δύο ανεξάρτητες ερευνητικές ομάδες ανακάλυψαν μεταλλάξεις του γονιδίου RET στη γαμετική σειρά οι οποίες έχουν αιτιολογική σχέση με το σύνδρομο ΜΕΝ2Α (Mulligan et al 1993) και το οικογενές ΜΚΘ (Donis-Keller et al 1993). Ένα χρόνο αργότερα το ΜΕΝ2Β συνδέθηκε επίσης με μεταλλάξεις του γονιδίου RET (Eng et al 1994, Carlson et al 1994, Hofstra et al 1994).
Στο σύνδρομο ΜΕΝ 2Α η πλειοψηφία των μεταλλάξεων εντοπίζεται στην πλούσια σε κυστεΐνες εξωκυττάρια περιοχή (στα κωδικόνια 609, 611, 618, 620 του εξωνίου 10 και στο κωδικόνιο 634 του εξωνίου 11). Είναι ενδιαφέρον ότι το 85% των μεταλλάξεων παρουσιάζεται στο εξώνιο 11 στο κωδικόνιο 634 που είναι και η συχνότερη θέση (Elisei 2008).
Στο σύνδρομο ΜΕΝ 2Β το 95% των περιπτώσεων συνδέεται με μία σημειακή μετάλλαξη στο κωδικόνιο 918 του εξωνίου 16.
Μετά από την ευρεία εφαρμογή της γενετικής ανάλυσης του γονιδίου RET στους ασθενείς με ΜΕΝ2 και με την παράλληλη ανάπτυξη της τεχνολογίας έχει γίνει δυνατή η ευρεία και γρήγορη ανίχνευση των μεταλλάξεων RET (Εικόνα 2). Πρόσφατα έχουν βρεθεί μεταλλάξεις και στα εξώνια 5, 8, 13, 14 και 15. Έτσι αυξήθηκαν και αυξάνονται συνεχώς τόσο ο αριθμός όσο και οι τύποι νέων μεταλλάξεων που ευθύνονται για όγκους ΜΚΘ.
Υπάρχει μεγάλη ποικιλία διεθνώς τόσο στο προφίλ όσο και την κατανομή των μεταλλάξεων από χώρα σε χώρα.

Εικόνα 2. Σχηματική παράσταση του γονιδίου ret (21 εξώνια) και των θέσεων στις οποίες έχουν περιγραφεί οι συχνότερες μεταλλάξεις στα οικογενή ΜΚΘ (FMTC, MEN2A, MEN2B) και στο συγγενές σύνδρομο Hirschprung (HSCR). CLΑ: Cutaneous Lichen Amyloidosis (De Groot et al. 2006).
Στη χώρα μας εμφανίζονται μέχρι σήμερα οι μεταλλάξεις που απεικονίζονται στον Πίνακα 1. Τα δεδομένα αυτά είναι υπό δημοσίευση από το κέντρο αναφοράς για τα ΜΕΝ2 της Θεραπευτικής Κλινικής του Παν. Αθηνών και του 1ου Ενδοκρινολογικού Τμ. του Νοσ. «ΑΛΕΞΑΝΔΡΑ».
Είναι ενδιαφέρον ότι το μεγαλύτερο ποσοστό των ασθενών δηλαδή ένα 36,21% φέρουν την μετάλλαξη TGG/TGC στο κωδικόνιο 533 του εξωνίου 8 η οποία είναι πολύ σπάνια σε άλλες χώρες όπως φαίνεται από την διεθνή βιβλιογραφία (Sarika et al 2012).
ΕΞΩΝΙΟ ΚΩΔΙΚΟΝΙΟ ΜΕΤΑΛΛΑΞΗ ΠΟΣΟΣΤΟ
8 533 TGG/TGC 36, 21 %
10 618 TGC/CGC 3, 45 %
10 618 TGC/TAC 1, 72 %
10 618 TGC/TCC 1, 72 %
10 620 TGC/TAC 1, 72 %
10 620 TGC/CGC 5, 18 %
10 620 TGC/TTC 1, 72 %
11 634 TGC/CGC 18, 97 %
11 634 TGC/TAC 12, 07 %
11 634 TGC/TTC 1, 72 %
13 768 GAG/GAC 1, 72 %
14 804 GTG/ATG 5, 18 %
16 918 AGC/ATG 8, 62 %
Πίνακας 1. Κατανομή μεταλλάξεων RET στα οικογενή ΜΚΘ σε ένα κέντρο αναφοράς στην Ελλάδα.
4. Σχέση Γονοτύπου Φαινότυπου
Μελέτη μεγάλων σειρών κληρονομούμενων ΜΚΘ έδειξε σημαντική συσχέτιση μεταξύ συγκεκριμένων μεταλλάξεων RET τόσο με το φαινότυπο δηλαδή τα κλινικά χαρακτηριστικά της νόσου (MEN2A, MEN2B,FMTC) όσο και με την επιθετικότητα του ΜΚΘ. Συγκεκριμένα η μετάλλαξη Μ918Τ που χαρακτηρίζει το σύνδρομο ΜΕΝ2Β είναι η πιο επιθετική και η πλειοψηφία των ασθενών με αυτήν την μετάλλαξη παρουσιάζουν αυξημένη θνητότητα σε νέα ηλικία. Αντίθετα ασθενείς που φέρουν μεταλλάξεις όπως Y791F και A883T είναι πιθανόν να μην αναπτύξουν ποτέ ΜΚΘ στην διάρκεια της ζωής τους (Elisei et al 2008).
Το είδος της μετάλλαξης συσχετίζεται τόσο με τη ηλικία ανάπτυξης του ΜΚΘ όσο και με την διαβάθμιση της επικινδυνότητας της νόσου (risk levels) η οποία κατευθύνει και τον χειρισμό της (Kouvaraki et al 2005) (Εικόνα 3).
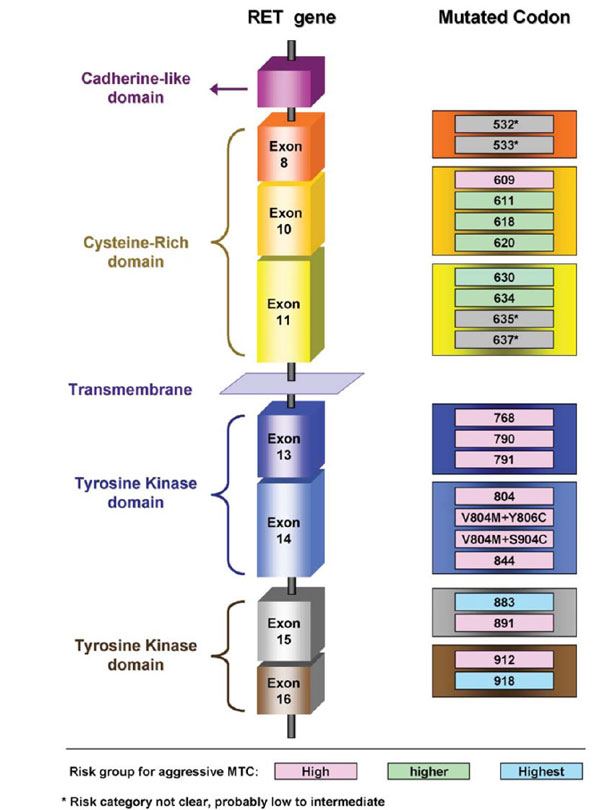
Εικόνα 3. Ομάδες επικινδυνότητας ανάλογα με την μετάλλαξη του γονιδίου RET σε οικογενή σύνδρομα ΜΚΘ (Kouvaraki et al 2005)
Η Αμερικανική Θυρεοειδική Εταιρεία (ΑΤΑ) έχει κατατάξει την βαρύτητα της νόσου ανάλογα με τα επίπεδα επικινδυνότητας και προτείνει την κατάλληλη αντιμετώπιση Πίνακας 2.
Πίνακας 2. Επίπεδα επικινδυνότητας και συνιστώμενη ηλικία για έλεγχο και προφυλακτική θυρεοειδεκτομή από την Αμερικανική Θυρεοειδική Εταιρεία (ΑΤΑ).
|
Επίπεδο κινδύνου |
Ηλικία για έλεγχο RET |
Ηλικία πρώτου υπέρηχου |
Ηλικία ελέγχου πρώτης καλσιτονίνης |
Ηλικία προφυλακτικής θυρεοειδεκτομής |
|
Δ |
Το συντομότερο στο 1ο έτος ζωής |
Το συντομότερο στο 1ο έτος ζωής |
Στον 6ο μήνα εάν δεν έχει προηγηθεί θυρεοειδεκτομή |
Το συντομότερο στο 1ο έτος ζωής |
|
Γ |
<3-5 έτη |
<3-5 έτη |
<3-5 έτη |
Πριν το 5ο έτος |
|
Β |
<3-5 έτη |
<3-5 έτη |
<3-5 έτη |
Μπορεί και μετά το 5ο έτος με αυστηρά κριτήρια |
|
Α |
<3-5 έτη |
<3-5 έτη |
<3-5 έτη |
Μπορεί και μετά το 5ο έτος με αυστηρά κριτήρια |
5. Μοριακές μέθοδοι για ανάλυση ΜΚΘ
Έχουν χρησιμοποιηθεί πολλές μέθοδοι μοριακής ανάλυσης από το 1993 που ξεκίνησε ο μοριακός έλεγχος για τους ασθενείς με ΜΚΘ.
Σήμερα τα περισσότερα εργαστήρια εφαρμόζουν την άμεση ανάλυση αλληλουχίας του γονιδίου (direct sequencing) ξεκινώντας από τα πιο συνήθη εξώνια και επί αρνητικών επεκτείνεται ο έλεγχος στα σπανιότερα εξώνια (Εικόνες 4,5,6).
Για τον έλεγχο του κωδικόνιου 918 στο εξώνιο 16, εφόσον ο ασθενής έχει τον χαρακτηριστικό φαινότυπο του συνδρόμου ΜΕΝ2Β χρησιμοποιείται ακόμη και η κατάτμηση με περιοριστικό ένζυμο η οποία είναι απολύτως ειδική, πολύ συντομότερη και οικονομικότερη (Εικόνα 7). Στην εικόνα 1 αποτυπώνεται μια ανάλυση αλληλουχίας DNA σε γέλη ηλεκτροφόρησης σε ακρυλαμίδη ενός ασθενή με μετάλλαξη στο εξώνιο 11 και ενός φυσιολογικού μάρτυρα. Στην εικόνα 2 αποτυπώνεται το ηλεκτρογράφημα μια αλληλουχίας DNA από αυτόματο αναλυτή αλληλούχησης DNA ενός φυσιολογικού δείγματος και στην εικόνα 3 το αντίστοιχο ηλεκτρογράφημα ενός ασθενούς ο οποίος φέρει την μετάλλαξη στο εξώνιο 8 του γονιδίου RET.
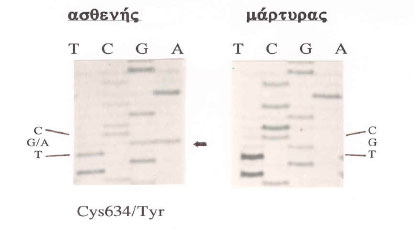
Εικόνα 4.
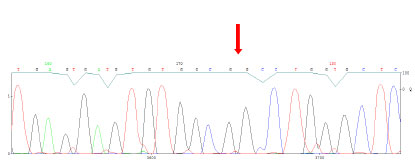


Εικόνα 7.
6. Αναγκαιότητα γενετικής ανάλυσης
Το σύνδρομο ΜΕΝ2 είναι το καλύτερο παράδειγμα για την αποτελεσματικότητα της μοριακής διαγνωστικής στην ογκολογία δεδομένου ότι προσφέρεται δυνατότητα ίασης σε προκλινικό στάδιο (Cerrato et al Review 2009).
Ο γενετικός έλεγχος στους ασθενείς με ΜΚΘ είναι αναγκαίος για τον χαρακτηρισμό των κληρονομούμενων μορφών της νόσου. Πρόσφατα έχουν δημοσιευτεί κατευθυντήριες οδηγίες για τον γενετικό έλεγχο ατόμων με ΜΚΘ από την Ευρωπαϊκή Θυρεοειδική Εταιρεία (European Thyroid Journal 2012 Elisei et al).
Εφόσον εντοπιστεί μετάλλαξη στην οικογένεια στην συνέχεια ελέγχονται τα κληρονομικώς συνδεόμενα μέλη της για τον εντοπισμό πιθανών φορέων για την έγκαιρη διάγνωση και προληπτική αντιμετώπιση τους. Τα μέλη της οικογένειας τα οποία βρίσκονται αρνητικά ως προς την μετάλλαξη απαλλάσσονται από περαιτέρω έλεγχο.
Εάν δεν βρεθεί μετάλλαξη στον ασθενή που πάσχει από ΜΚΘ και δεν υπάρχει γνωστό οικογενειακό ιστορικό, οι συγγενείς παρακολουθούνται με προσδιορισμό των επιπέδων καλσιτονίνης. Η πιθανότητα όμως να πρόκειται για κληρονομούμενη μορφή με φυσιολογικό γονίδιο ret είναι πολύ μικρή.
Η γενετική ανάλυση είναι αναγκαία σε όλους τους ασθενείς με ΜΚΘ για τον εντοπισμό πιθανών οικογενών περιπτώσεων ακόμη και σε ασθενείς με φαινομενικά σποραδικό ΜΚΘ.
Ο χαρακτηρισμός της μετάλλαξης οδηγεί στην καλύτερη εκτίμηση πρόγνωσης και στην επιλογή κατάλληλης θεραπευτικής αγωγής για τον πάσχοντα. Η μεγαλύτερη αξία όμως αφορά τους προγόνους και τους συγγενείς πρώτου βαθμού στους οποίους προσφέρεται η δυνατότητα προφυλακτικής θυρεοειδεκτομής αφ’όσον βρεθούν φορείς της μετάλλαξης.
Βιβλιογραφία
- Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, Jackson CE, Wells SA, Godfellow PJ, Donis-Keller H 1994 Single missence mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci USA 91(4):1579-1583.
- Cerrato A, De Falco, Santro M. 2009 Molecular genetics of medullary thyroid carcinoma: the quest for novel therapeutic targets. J of Mol Endocrinol 43:143-15.
- Donis-Keller H, Dou S, Chi D, Carlson KN, Toshima K, Lairmore TC, Howe JR, Moey JF, Goodfellow P, Wells SA 1993 Mutations in the RET proto-oncogene are associated with MEN2A and FMTC. Hum Mol Genet 2(7):851-856.
- Elisei R, Romei C, Cosci B, Molinaro E, Agate L, Bottici V, Pinchera A. 2008 RET point mutations in Thyroid Carcinoma. Atlas of Genetics and Cytogenetics in Oncology and Haematology April 2008.
- Elisei R, Alevizaki M, Conte-Devolx B, Frank-Raue K, Leite V, Williams GR 2012. 2012 European Thyroid Association Guidelines for Genetic Testing and Its Clinical Consequences in Medullary Thyroid Cancer Eur Thyroid J 1:216–231.
- Eng C, Smith DP, Mulligan LM, Nagai MA, Healey CS, Ponder MA, Gardner E, Scheumann GF, Jackson CE, Tunnacliffe A et al 1994 Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet 3(2):237-241.
- Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel R et al 1996 The Relationship Between Specific RET Proto-oncogene Mutations and Disease Phenotype in Multiple Endocrine Neoplasia Type 2. JAMA 19:1575-1579.
- De Groot JW, Links T, Plukker JT, Lips C, Hofstra RM. 2006 RET as a Diagnostic and Therapeutic Target in Sporadic and Hereditary Endocrine Tumors.Endocrine Reviews 27(5):535-560.
- Hazard JB, Hawk WA, Grile G, Jr 1959 Medullary (solid) carcinoma oh the thyroid; a clinicopathologic entity. J Clin Endocrinol Matab 19:152-161.
- Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, Pasini B, Hoppener JW, van Amstel HK, Romeo G, et al 1994 A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 27; 367(6461):375-376.
- Kloos R, Eng C, Evans D, Francis G, Gagel R, Gharib H, Moley J, Pacini F, Ringel M, Schlumberger M and Wells S 2009 Medullary Thyroid Cancer:
- Management Guidelines of the American Thyroid Association. Thyroid19:565-612.
- Kouvaraki MA, Shapiro SE, Perrier ND, Cote GJ, Gagel RF, Sherman SI, Lee JE, Evans DB 2005 RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 15(6):531-544.
- Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, et al 1993 Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363(6428):458-460
- Sarika HL, Papathoma A, Garofalaki M, Vasileiou V, Vlassopoulou B, Anastasiou E, Alevizaki M. 2012 High prevalence of exon 8 g533c mutation in apparently sporadic medullary thyroid carcinoma (mtc) in Greece). Clin Endocrinol 77(6):857-62.
Created: October 25, 2014
Last update: October 25, 2014