
Αστερούλα Καϊμάρα-Παπαθανασίου
Παιδοενδοκρινολόγος
τ. Διευθύντρια Ενδ. Τμήματος Νοσοκομείου Παίδων Π & Α. Κυριακού, Αθήνα
Αλεξάνδρα Χρυσουλίδου
Ενδοκρινολόγος
Επιμελήτρια Α’, Ενδοκρινολογικό Τμήμα, Θεαγένειο Νοσοκομείο, Θεσσαλονίκη
Ελένη Κούστα
Ενδοκρινολόγος
Κέρκυρα
1. Eισαγωγή
Η φυσιολογική αύξηση του ανθρώπου είναι το αποτέλεσμα μιας πολύπλοκης διαδικασίας, η οποία προκύπτει από την αλληλεπίδραση πολλαπλών γονιδίων σημαντικων για την εξελιξη και επιβίωση του ανθρώπινου είδους (Rosenfeld 2003). Κύριος ρυθμιστής της διαδικασίας αυτής είναι το κεντρικό νευρικό σύστημα (ΚΝΣ), το οποίο ρυθμίζει την έκκριση της αυξητικής ορμόνης (GH) και την δράση των IGFs (ινσουλινόμορφων αυξητικών παραγόντων, insulin like growth factors), των πρωτεϊνών που δρουν στο τελικό όργανο.
Ο ακέραιος άξονας GH-IGF1 αποτελεί προϋπόθεση για φυσιολογική ενδομήτρια και εξωμήτρια αύξηση.
Στο παρακάτω κείμενο θα αναφερθούν: α. η λειτουργία του άξονα GH-IGF1 ως προς την ρύθμιση της αύξησης και β. οι διαταραχές του άξονα GH-IGF1, οι οποίες προκαλούν καθυστέρηση της αύξησης και χαμηλό ανάστημα. Οι διαταραχές αυτές περιλαμβάνουν νοσήματα που οφείλονται σε ανεπαρκή έκκριση αυξητικής ορμόνης (growth hormone deficiency, GHD) και ανεπαρκή ανταπόκριση (αντίσταση) στη δράση της GH (growth hormone insensitivity, GHI).
Περιγράφεται ο ορισμός, η διάγνωση και η αντιμετώπιση των σημαντικότερων διαταραχών.
1.1. Ο άξονας GH-IGF1
Η GH είναι ο κύριος ρυθμιστής της κατά μήκος αύξησης του ανθρώπου. Οι πολλαπλές δράσεις της GH στην αύξηση και τον μεταβολισμό διαμεσολαβούνται από το σύστημα των IGFs και είναι είτε IGF -εξαρτώμενες είτε IGF-ανεξάρτητες (άμεσες) (Savage M. 2010).
Το σύστημα των IGF περιλαμβάνει τoν IGF1, IGF2, τις συνδέουσες πρωτεΐνες (IGFBPs, insulin like binding proteins) και τον υποδοχέα των IGF ( IGFR).
Οι IGFs κυκλοφορούν στον ορό συδεδεμένες με τις IGFBPs ,οι οποίες βοηθούν στην επιμήκυνση του χρόνου ημίσειας ζωής των IGFs, την μεταφορά τους στα όργανα- στόχος και την αλληλεπίδρασή τους με τους υποδοχείς (Cohen 2006).
Η σύνδεση της GH με τον υποδοχέα της (GHR) ενεργοποιεί ένα σύνθετο ενδοκυττάριο σηματοδοτικό καταρράκτη, ο οποίος οδηγεί στην μεταγραφή πολλών γονιδίων, συμπεριλαμβανομένου του γονιδίου του IGF1. Ο IGF1 εκκρίνεται από το ήπαρ και κυκλοφορεί με την μορφή τριμερούς ένωσης (ternary complex) 150 kDa μαζί με δύο πρωτεϊνες εξαρτώμενες από την GH, την IGFBP3 και την ALS (acid labile subunit).
Ο IGF1 διαμεσολαβεί τις περισσότερες δράσεις της GH και προκαλεί αρνητική παλίνδρομη ρύθμιση (negative feed back) στο επίπεδο της υπόφυσης. Η ακεραιότητα του τριμερούς IGF1-IGFBP3-ALS στον ορό είναι απαραιτητη για την φυσιολογικη λειτουργία του συστήματος των IGFs (Domene 2004).
Εκτός από τον IGF1 ηπατικής προέλευσης που κυκλοφορεί στον ορό, έχει ιδιαίτερη σημασία ο τοπικά παραγóμενος IGF1. Είναι ενδιαφέρον ότι η αύξηση των μυών που έχουν υποστεί καταστροφή του IGF1 (IGF1 knockout mice) είναι σχεδόν φυσιολογική παρά τα πολύ χαμηλά επίπεδα IGF1 στην κυκλοφορία, παρατήρηση που αποδίδεται στον τοπικά παραγόμενο IGF1. Επιπλέον ασθενείς με έλλειψη ALS δεν παρουσιάζουν σοβαρή καθυστέρηση στην αύξηση, πιθανόν λόγω διατήρησης των τοπικά παραγομένων IGFs.
Η ενδομήτρια αύξηση ρυμίζεται κυρίως από την ύπαρξη αποθεμάτων θρεπτικών συστατικών, τα οποία επηρεάζουν την σύνθεση IGF1 και IGF2. Η εκλεκτική καταστροφή των γονιδίων IGF1 και IGF2 σε μύες οδηγεί σε ελάττωση της κατά μήκος αύξησης κατά 40% (Lupu 2001).
Επίσης, μεταλλάξεις του γονιδίου του IGF1 ή του IGFR προκαλούν καθυστέρηση της ενδομήτριας αύξησης στον άνθρωπο (Woods 1996, Wallenkamp 2006).
Η αύξηση κατά την εξωμήτρια ζωή εξαρτάται από την επίδραση της GH στον υποδοχέα της και την διέγερση της παραγωγής του IGF1. Σε περιπτώσεις συγγενούς ανεπάρκειας GH από γενετικά αίτια ή λόγω διαταραχών του υποδοχέα της GH με επακόλουθο ανεπάρκεια IGF1, οι ασθενείς παρουσιάζουν ενωρίς σοβαρού βαθμού καθυστέρηση της αύξησης (Dattani 2005).
Το σύστημα των IGFs έχει πολλαπλές δράσεις σε όλα τα συστήματα του ανθρώπινου οργανισμού και δεν σχετίζεται μόνο με τη ρύθμιση της αύξησης. Ο IGF1 ευαισθητοποιεί την ινσουλίνη αφενός μέσω της καταστολής της έκκρισης GH και αφετέρου δρώντας άμεσα σε περιφερικούς ιστούς. Έρευνες των τελευταίων ετών αποκαλύπτουν την δράση του συστήματος των IGFs στην διαφοροποίηση και τον πολλαπλασιασμό των νευρικών κυττάρων και την πρόληψη της απόπτωσης με αποτέλεσμα την διατήρηση της επιβίωσής τους (Russo 2005). Επίσης από επιδημιολογικές μελέτες φαίνεται ο σημαντικός ρόλος του συστήματος στην καρκινογένεση, όπου τα αυξημένα επίπεδα IGF1 συσχετίζονται με την εμφάνιση ορισμένου τύπου κακοήθων νοσημάτων. Οι κλινικές μελέτες σε παιδιά που παίρνουν θεραπεία υποκαταστάσεως με IGF1 δεν αναφέρουν αυξημένη επίπτωση καρκίνου. Φαίνεται, λοιπόν, ότι η χορήγηση IGF1 σε ασθενείς, η οποία στοχεύει στην υποκατάσταση και διατήρηση των επιπέδων του IGF1 σε φυσιολογικά επίπεδα, είναι ασφαλής .
1.2. Το συνεχές φάσμα των διαταραχών του άξονα GH- IGF
Η λειτουργία του συστήματος GH-IGF1 μπορεί να διαταραχθεί σε οποιοδήποτε σημείο του άξονα, από βλάβη που αφορά, είτε παράγοντες σχετιζόμενους με την έκκριση της GH, είτε παράγοντες σχετιζόμενους με την φυσιολογική παραγωγή και δράση του IGF1. Η προκληθείσα διαταραχή αντανακλά στο αμέσως κατώτερο επίπεδο, με αποτέλεσμα διαταραχή των γονιδίων και πρωτεϊνών και ορμονική δυσλειτουργία που εκδηλώνεται με καθυστέρηση της αύξησης και πιθανόν δυσμορφικά σημεία.
Η έλλειψη IGF1 οδηγεί σε καθυστέρηση ανάπτυξης και μπορεί να οφείλεται είτε σε έλλειψη GH (συνεπώς ανεπαρκή παραγωγή IGF1) είτε σε διαταραχή της σύνδεσης της GH με τον υποδοχέα της ή της ενδοκυττάριας μετάδοσης του σήματος, όπου η παραγωγή GH είναι μεν φυσιολογική αλλά δεν παράγεται IGF1. Οι ασθενείς με έλλειψη GH έχουν παρόμοια κλινική εικόνα ασχέτως αιτιολογίας, είτε η έλλειψη GH οφείλεται σε σοβαρή αντίσταση στην GH, συγγενή έλλειψη GH ή σοβαρού βαθμού έλλειψη IGF1.
Επομένως το φάσμα των διαταραχών του άξονα είναι ευρύ και περιλαμβάνει στο ένα άκρο την σοβαρή έλλειψη GH (διαταραχή έκκρισης) και στο άλλο άκρο την σοβαρή αντίσταση στη δράση της GH (διαταραχή ευαισθησίας) (Cohen 2006).
Στα ενδιάμεσα σημεία βρίσκονται ασθενείς με ηπιότερη καθυστέρηση της αύξησης,οι οποίοι ανήκουν στην ομάδα του ιδιοπαθούς χαμηλού αναστήματος (ISS).
Η κατηγορία αυτή περιλαμβάνει εκτός από τα παιδιά με φυσιολογική GH πιθανόν και παιδιά με ήπια ανεπάρκεια GH καθώς και παιδιά με μικρού βαθμού αντίσταση στην GH.
Το κοινό χαρακτηριστικό όλων των ασθενών είναι η έλλειψη IGF1, η οποία κυμαίνεται ανάλογα με τον βαθμό της διαταραχής.
Η παρουσίαση του φάσματος των διαταραχών κατ’αυτόν τον τρόπο (σχήμα 1) είναι σχετικά αυθαίρετη και πιθανόν να τροποποιηθεί μελλοντικά με την πρόοδο στην γενετική διερεύνηση των μοριακών μηχανισμών, βοηθά όμως στην κατανόηση αυτών των καταστάσεων.
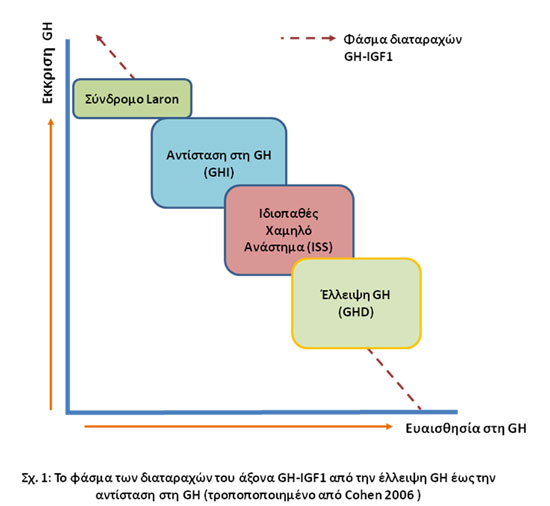
2. Ορισμός της ‘έλλειψης IGF1’ στο πλαίσιο των διαταραχών του άξονα GH-IGF
Το σύστημα GH-IGF1 μπορεί να θεωρηθεί αντίστοιχο με άλλα ενδοκρινικά συστήματα, όπου υπάρχει μια κεντρική, τροφική ορμόνη (GH) και μια περιφερικά δραστική ορμόνη (IGF1).
Η διαταραχή στην έκκριση της GH έχει σαν αποτέλεσμα την ελάττωση των επιπέδων του IGF1 και αντιστρόφως η ελαττωμένη παραγωγή IGF1 οδηγεί σε αύξηση της έκκρισης της GH (Ranke M 2006).
Επομένως, ελάττωση των επιπέδων του IGF1 οφειλόμενη σε ελαττωμένη έκκριση GH μπορεί να ονομαστεί “δευτεροπαθής έλλειψη IGF1”.
Αντίστοιχα, η ελάττωση της παραγωγής του IGF1 η οποία δεν οφείλεται σε μειωμένη έκκριση GH μπορεί να ονομαστεί “ πρωτοπαθής έλλειψη IGF1”.
Η ονοματολογία αυτή είναι κατατοπιστική, παρότι στην πραγματικότητα δεν ισχύει πλήρης αντιστοιχία μεταξύ έλλειψης IGF1 και έλλειψης GH, όπως στα άλλα ενδοκρινικά συστήματα. Η ταύτιση ισχύει μόνο στον φαινότυπο της σοβαρής διαταραχής έλλειψης GH και σοβαρής έλλειψης IGF1 οφειλόμενης σε αντίσταση στο επίπεδο των υποδοχέων ή στα αρχικά στάδια της μεταγωγής του ενδοκυτταρίου σήματος μέσω JAK-STAT μετά την πρόσδεση της GΗ στον υποδοχέα (Ranke 2006).
Στην πραγματικότητα υπάρχει ευρύ φάσμα φαινοτύπων, το οποίο οφείλεται στις πολυάριθμες ποιοτικές και ποσοτικές διαταραχές του άξονα ( Woods 1997).
2.1. Έλλειψη GH
Χαρακτηρίζεται από ελαττωμένη σύνδεση GH-GHR, με συνέπεια την ελλειπή μετάδοση του σήματος για μεταγραφή του γονιδίου του IGF1 και επομένως ανεπαρκή παραγωγή IGF1, IGFBP3 και ALS.
Τα επίπεδα του IGF1 είναι χαμηλά και συσχετίζονται θετικά με την μέγιστη τιμή στις δοκιμασίες διέγερσης GH, ενώ σε ήπιες μορφές GHD και ιδιοπαθούς χαμηλού αναστήματος (ISS) ,τα επίπεδα IGF1 αλληλοεπικαλύπτονται.
2.2. Ιδιοπαθές χαμηλό ανάστημα (ISS)
Περιλαμβάνει ασθενείς με καθυστέρηση αύξησης και φυσιολογική GH.
Tο ύψος είναι < -2 SD, η ταχύτητα αύξησης είναι φυσιολογική ή χαμηλή, το βάρος γεννήσεως φυσιολογικό και δεν υπάρχουν ενδοκρινοπάθειες ή άλλα χρόνια νοσήματα.
Η καθυστέρηση της αύξησης οφείλεται σε πληθώρα αιτίων, μερικά από τα οποία αφορούν διαταραχές του άξονα GH-IGF1 (Wit 2008).
Ετερόζυγες μεταλλάξεις του υποδοχέα της GH έχουν αναφερθεί σαν πιθανή αιτία του ιδιοπαθούς χαμηλού αναστήματος. Δεν είναι βέβαιο ότι οι μεταλλάξεις αυτές προκαλούν χαμηλό ανάστημα, καθώς, όπως αναφέρεται σε μελέτη κατοίκων του Ισημερινού, δεν υπάρχει διαφορά ύψους στους ενηλίκους με ετερόζυγο μετάλλαξη Ε180 σε σύγκριση με φυσιολογικά άτομα χωρίς μετάλλαξη (Guevarra-Aguirre 2007, Savage 2006).
Περίπου 25-50% των παιδιών με ιδιοπαθές χαμηλό ανάστημα έχουν επίπεδα IGF1 < -2 σταθερές αποκλίσεις (SDS ). Τα χαμηλά επίπεδα IGF1 σε συνδυασμό με την φυσιολογική έκκριση GH υποδηλώνουν ότι πιθανόν να υπάρχει μερική αντίσταση στην GH, η οποία συμβάλλει στην καθυστέρηση της αύξησης.
Οι χαμηλές τιμές IGF1 μπορεί να οφείλονται και σε άλλα αίτια, όπως ελλειπή θρέψη, συστηματικά νοσήματα ή απλή καθυστέρηση ύψους και εφηβείας. Εάν τα αίτια αυτά αποκλεισθούν, είναι πιθανόν οι ασθενείς που έχουν χαρακτηρισθεί ως ασθενείς με ιδιοπαθές χαμηλό ανάστημα να παρουσιάζουν πρωτοπαθή έλλειψη IGF1.
2.3. Αντίσταση στην GH (GH resistance, GH insensitivity)
Περιλαμβάνει ασθενείς με φυσιολογική ή αυξημένη GH στον ορό και χαμηλά επίπεδα IGF1. Παρά το γεγονός ότι ο IGF1 ανευρίσκεται πάντοτε χαμηλός, υπάρχει ποικιλομορφία στον φαινότυπο που περιλαμβάνει από ήπια καθυστέρηση αύξησης έως σοβαρού βαθμού χαμηλό ανάστημα,αλλά και στα βιοχημικά χαρακτηριστικά ανάλογα με τη βαρύτητα της διαταραχής.
Η ομάδα αυτή περιλαμβάνει
Α.Μεταλλάξεις του υποδοχέα της GH (GHR)
B. Διαταραχές της μεταγωγής του σήματος της GH (μεταλλάξεις μετά τον υποδοχέα (post-GH receptor)
Γ.Διαταραχές του IGF1 και του υποδοχέα του (IGF1R)
Η αναλυτική αιτιολογική ταξινόμηση των διαταραχών της έλλειψης IGF1 φαίνεται στον Πίνακα 1.
3. Διερεύνηση ασθενών με διαταραχές του άξονα GH-IGF1 (πρωτοπαθής έλλειψη IGF1)
Η λεπτομερής κλινική εξέταση και η εκτίμηση των αυξολογικών παραμέτρων (ύψους, βάρους, ταχύτητας αύξησης) είναι σημαντικές για να αποφασισθεί αν το παιδί χρειάζεται περαιτέρω διερεύνηση.
Εαν η ταχύτητα αύξησης δεν είναι φυσιολογική και ο IGF1 είναι χαμηλός, υπάρχει υποψία έλλειψης GH. Εφόσον αποκλεισθεί υποθυρεοειδισμός και άλλα αίτια καθυστέρησης ανάπτυξης, απαιτούνται 2 δοκιμασίες διεγέρσεως με επίπεδα GH< 10 ng/ml για την επιβεβαίωση της διάγνωσης GHD καθώς και MRI υποθαλάμου-υποφύσεως . Σε παιδιά μικρότερα των 3 ετών τα χαμηλά επίπεδα IGFBP3 συμβάλλουν στην επιβεβαίωση της διάγνωσης.
Εάν η ταχύτητα αύξησης είναι χαμηλή, ο IGF1 επίσης χαμηλός, αλλά η GH φυσιολογική, πρόκειται για αντίσταση στην GH.
H περαιτέρω διερεύνηση της αιτίας απαιτεί τον προσδιορισμό IGFBP3, ALS και GHBP. Ο λόγος IGF1 προς IGFBP3 καθορίζει την βιοδιαθεσιμότητα του IGF1.
Οι διαταραχές λόγω μεταλλάξεων των STAT5b και ALS συνοδεύονται από σοβαρού βαθμού έλλειψη IGF1, IGFBP3 και ALS, ενώ η GHBP είναι φυσιολογική (Rosenfeld 2007, Domene 2009). Ασθενείς με μεταλλάξεις του IGF1R έχουν φυσιολογικά ή αυξημένα επίπεδα IGF1 (Walenkamp 2008) (Πίνακας 2).
Πίνακας 2. Κλινικά και βιοχημικά χαρακτηριστικά ασθενών με διαταραχές του άξονα GH-IGF1.
| Μετάλλαξη γονιδίου | Βάρος Γέννησης | Εξωμήτρια αύξηση | GH | GHBP | IGF1 | IGFBP3 | ALS | Κλινικά χαρακτηριστικά |
| GHR (εξωκυττάριος) | φ | Σοβαρή καθυστέρηση | £ | ¤ | ¤ | ¤ | ¤ | Κρανιοπροσωπική δυσμορφία |
| GHR (διαμεμβρανικός) | φ | Σοβαρή καθυστέρηση | £ | φ ή £ | ¤ | ¤ | ¤ | Κρανιοπροσωπική δυσμορφία |
| GHR (ενδοκυττάριος) | φ | Σοβαρή καθυστέρηση | £ | φ | ¤ | ¤ | ¤ | Κρανιοπροσωπική δυσμορφία |
| φ | Σοβαρή καθυστέρηση | £ | ¤ | ¤ | ¤ | νοσοανεπάρκεια, £ Προλακτίνη |
||
| ALS | φ | Σοβαρή καθυστέρηση | £ | φ | ¤ | ¤ | ¤ | Αντίσταση στην ινσουλίνη |
| IGF1 | ¤ | Σοβαρή καθυστέρηση | £ | φ | ¤ | φ | φ | Κώφωση, μικροκεφαλία, πνευματική καθυστέρηση, καθυστέρηση ήβης |
| Βιοανενεργός IGF1 | ¤ | Σοβαρή καθυστέρηση | £ | φ | φ ή £ | φ | φ | Κώφωση, μικροκεφαλία, πνευματική καθυστέρηση |
| IGF1R | ¤ | Σοβαρή καθυστέρηση | £ | φ | φ ή £ | φ | φ | Μικροκεφαλία, διαταραχές σίτισης |
Φ= φυσιολογικό
To IGF generation test. Το σύνδρομο Laron χαρακτηρίζεται από πρωτοπαθή βλάβη στην παραγωγή του IGF1, λόγω διαταραχής του υποδοχέα της GΗ. Η κλινική εικόνα ομοιάζει με αυτήν της σοβαρής έλλειψης αυξητικής ορμόνης (GHD), με χαμηλές IGF1 /IGFBP3, αλλά παρατηρείται αδυναμία ανταπόκρισης των επιπέδων του IGF1 και της αύξησης στην εξωγενώς χορηγούμενη GH.
Η αντίσταση στην δράση της GH στην περίπτωση αυτή αποτελεί μια ειδική περίπτωση πρωτοπαθούς έλλειψης IGF1, η οποία μπορεί να ανταποκριθεί μόνο στη χορήγηση IGF1.
Η σπουδαιότερη λειτουργική δοκιμασία για την διάγνωση της αντίστασης στην GH (GH insensitivity syndrome, GHIS) είναι το IGF generation test.
Η δοκιμασία συτή βασίζεται στην ικανότητα του οργανισμού να αυξάνει την παραγωγή IGF1 και των GH-εξαρτώμενων πεπτιδίων μετά από εξωγενή χορήγηση GH. Συνίσταται στην υποδόρια ένεση 33 μg/kg GH ημερησίως επι 4 ημέρες και τον προσδιορισμό των επιπέδων IGF1 και IGFBP3 πριν και μετά το τέλος της δοκιμασίας. Εχει χρησιμοποιηθεί τα τελευταία 25 χρόνια, ως διαγνωστική δοκιμασία για την διερεύνηση παιδιών με χαμηλό ανάστημα, αλλά έχει και προγνωστική σημασία για την ανταπόκριση στη θεραπεία με GH.
Η δοκιμασία αυτή χρησιμοποιήθηκε αρχικά από τον Rudman το 1981 με τον σκοπό να εκτιμηθεί η βραχυπρόθεσμη ανταπόκριση στην θεραπεία με GH παιδιών με ιδιoπαθές χαμηλό ανάστημα. (Rudman 1981).
Αργότερα, ο Bloom λαμβάνοντας υπόψιν τις εως τότε γνώσεις, καθόρισε ως κριτήρια ανεπαρκούς ανταπόκρισης στην δοκιμασία χορήγησης GH την αύξηση του IGF1 λιγώτερο από 15 ng/ml και του IGFBP3 λιγώτερο από 400 ng/ml (Bloom 1994). Τα κριτήρια αυτά αναγνωρίζουν με βεβαιότητα ασθενείς με πραγματική αντίσταση (GHIS). Εντούτοις, η εμπειρία που αποκτήθηκε κατά τα επόμενα χρόνια σε ασθενείς με αποδεδειγμένη GHIS, GHD και ΙSS απέδειξε ότι η δοκιμασία δεν έχει 100% ειδικότητα.
Δηλαδή, η διαταραχή στην ευαισθησία στην GH δεν είναι αποκλειστικό γνώρισμα των ασθενών με κλασσική αντίσταση στην GH και μπορεί να παρατηρηθεί, τουλάχιστον ως ένα βαθμό, και σε παιδιά που ανήκουν στην κατηγορία του ιδιοπαθούς χαμηλού αναστήματος (ISS).
Εάν εφαρμοσθούν τα αρχικά αυστηρά κριτήρια μπορεί να διαγνωσθούν μόνο ασθενείς με σοβαρού βαθμού αντίσταση, ενώ ασθενείς που ανήκουν στις ‘γκρίζες ζώνες’ με ηπιότερου βαθμού αντίσταση, δυνατόν να μην διαγνωστούν δεδομένου ότι υπάρχει αλληλοεπικάλυψη μεταξύ των επιπέδων IGF1 και IGFBP3 των ασθενών με πλήρη έλλειψη, μερική αντίσταση και φυσιολογικών παιδιών) (Buckway 2001, Buckway 2002, Woods 1997).
Επειδή υπάρχει κάποια συσχέτιση μεταξύ της βραχυπρόθεσμης αύξησης του IGF1 και της μακροπρόθεσμης ανταπόκρισης στη θεραπεία με GH, το IGF generation test είναι ένα ευαίσθητο εργαλείο για την εκτίμηση της ευαισθησίας στην GH όχι μόνο στις σπάνιες και ακραίες μορφές αντίστασης του κλασσικού GHIS, αλλά και σε άλλες διαταραχές της αύξησης.
Oμως αφενός το test δεν έχει στανταριστεί επαρκώς όσον αφορά την δόση και τη διαρκεια χορήγησης της GH σε διεθνή πρωτόκολλα και αφετέρου δεν υπάρχουν επαρκή στοιχεία για να καθορισθούν τα φυσιολογικά όρια (normative data) στην ανταπόκριση στην GH. Συνεπώς, είναι τουλάχιστον προς το παρών, σε ορισμένες περιπτώσεις δύσκολη η ερμηνεία των αποτελεσμάτων.
Συνοψίζοντας, το test είναι αξιόπιστο για την διάγνωση της σοβαρής GHIS αλλά δεν είναι πολύ ισχυρό στην αποκάλυψη πιο ήπιων μορφών αντίστασης στην GH (Ranke 2006).
3.1. Μεταλλάξεις του υποδοχέα της GH ( GHR) (σύνδρομο Laron)
O υποδοχέας GHR είναι μία διαμεμβρανική πρωτεΐνη που ανήκει στην κατηγορία των υποδοχέων κυτοκινών (cytokine receptors) (Βackeljauw ). To υπεύθυνο γονίδιο για τον GHR εδράζεται στο χρωμόσωμα 5 και περιέχει 10 εξώνια. Τα εξώνια 3-7 κωδικοποιούν το εξωκυττάριο τμήμα του GHR, που επίσης αποτελεί και την δεσμευτική πρωτεΐνη της GH (GH binding protein, GHBP).
3.1.1. Γενετικές διαταραχές του GHR
H δράση της GH προϋποθέτει έναν λειτουργικό υποδοχέα GHR και ένα ακέραιο σηματοδοτικό μονοπάτι μετά από αυτόν. Oι γενετικές βλάβες που αφορούν τον GHR μπορεί να έχουν εντόπιση: α) στο εξωκυττάριο τμήμα του GHR (εξώνια 2-7) (απαλλείψεις και μεταλλάξεις σε αυτό το τμήμα προκαλούν το κλασσικό σύνδρομο Laron) και β) στο διαμεμβρανικό (εξώνιο 8) και ενδοκυττάριο τμήμα (εξώνια 9-10) του GHR (Walenkamp et al). Η κύρια διαφορά ανάμεσα στις δύο εντοπίσεις της γενετικής βλάβης είναι τα επίπεδα της GHBP: τα επίπεδα αυτά είναι χαμηλά στις βλάβες που αφορούν στο εξωκυττάριο τμήμα του GHR και φυσιολογικά ή υψηλά στις βλάβες που αφορούν στο διαμεμβρανικό και ενδοκυττάριο τμήμα του GHR.
Το 1966 ο Laron περιέγραψε μια οικογένεια με κλινική εικόνα σοβαρής έλλειψης αυξητικής ορμόνης, της οποίας τα μέλη είχαν υψηλές τιμές GH. Σήμερα γνωρίζουμε ότι η αιτία της σοβαρής αυτής διαταραχής ήταν αποτέλεσμα μοριακών διαταραχών του γονιδίου του υποδοχέα της GH. Η διαταραχή αυτή είναι γνωστή σαν σύνδρομο αντίστασης στην GH (GH insensitivity syndrome) ή σύνδρομο Laron. Οι διαταραχές του γονιδίου του GHR είναι οι συχνότερες γενετικές αιτίες πρωτοπαθούς έλλειψης IGF1, αλλά παραμένουν ακόμα σχετικά σπάνιες (έχουν δημοσιευθεί περίπου 300 περιπτώσεις στην παγκόσμια βιβλιογραφία) .
Κλινική εικόνα. Το κλασσικό σύνδρομο Laron περιλαμβάνει παιδιά με φυσιολογικό μήκος γέννησης, τα οποία όμως εμφανίζουν σοβαρή καθυστέρηση της αύξησης μετά τη γέννηση (μέσο ύψος – 6, εύρος -2 έως -12 SDS ) και τυπικά κλινικά χαρακτηριστικά (Laron 2004). Το προσωπείο περιλαμβάνει υποπλασία του μέσου τριτημορίου του προσώπου,προέχον μέτωπο και μπλέ σκληρούς χιτώνες των ματιών, μικρό πηγούνι , αραιά μαλλιά και μικρά άκρα. Στη νεογνική ηλικία παρατηρούνται συχνά υπογλυκαιμίες και εφίδρωση. Επίσης τα παιδιά εμφανίζουν κεντρικού τύπου παχυσαρκία και η αύξηση του βάρους τους επιδεινώνεται με το χρόνο φτάνοντας σε νοσογόνο παχυσαρκία κατά την ενήλικη ζωή. Η οδοντοφυία τους καθυστερεί και είναι ελαττωματική. Οι μύες του σώματος δεν είναι καλά αναπτυγμένοι, η σκελετική ωρίμανση αργεί, πιθανόν δε να υπάρχει και οστεοπενία. Τα γεννητικά όργανα είναι μικρά και η ήβη καθυστερεί , ωστόσο και τα δύο φύλα έχουν πλήρη σεξουαλική ωρίμανση και συνήθως δεν παρουσιάζουν προβλήματα τεκνοποίησης. Το μέσο τελικό ύψος κυμαίνεται μεταξύ 116-143 εκ. στους άνδρες και 108-136 εκ. στις γυναίκες, η δε πνευματική ανάπτυξη είναι φυσιολογική. Λόγω της παχυσαρκίας και της μειωμένης μυïκής ισχύος οι ασθενείς παρουσιάζουν ορθοπαιδικά προβλήματα και διαταραχές ύπνου. Παρά τα προβλήματά τους οι ασθενείς με σύνδρομο Laron ζουν πολλά χρόνια και συνήθως ξεπερνούν τα 70 έτη, ενώ δεν εμφανίζουν νοσηρότητα από καρκίνους, γεγονός το οποίο αποτελεί αντικείμενο μελέτης.
Ο φαινότυπος κυμαίνεται από ήπια διαταραχή της αύξησης χωρίς δυσμορφικά χαρακτηριστικά έως το κλασσικό σύνδρομο Laron. Υπάρχει μεγάλου βαθμού ετερογένεια στον φαινότυπο ακόμη και ανάμεσα σε ασθενείς με το κλασσικό σύνδρομο αντίστασης (GHIS). Παρά την απουσία συσχέτισης φαινοτύπου-γονοτύπου, παρατηρείται συσχέτιση μεταξύ των επιπέδων IGF1 και IGFBP3 και του SDS ύψους των ασθενών (Woods 1997).
Εργαστηριακά ευρήματα. Οι ασθενείς έχουν υψηλά επίπεδα GH και χαμηλά επίπεδα IGF-1( < 4 SD) και GHBP. Οι συνηθέστερες διαταραχές που αφορούν μεταλλάξεις στο εξωκυττάριο τμήμα συνοδεύονται από χαμηλά επίπεδα GHBP, ενώ εάν η βλάβη αφορά το ενδοκυττάριο τμήμα του υποδοχέα, πιθανόν τα επίπεδα GHBP να είναι φυσιολογικά. Τα επίπεδα IGFBP3 και ALS, ως εξαρτώμενα από την GΗ, είναι επίσης χαμηλά. Η δοκιμασία IGF-1 generation test μπορεί να βοηθήσει στην επιβεβαίωση της διάγνωσης και ο γενετικός έλεγχος οδηγεί στην ανεύρεση της υπεύθυνης μετάλλαξης του υποδοχέα της GH.
Γενετική βάση της διαταραχής. Το σύνδρομο μεταβιβάζεται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα, ενώ υπάρχουν σπάνιες περιπτώσεις μεταβίβασης με τον επικρατούντα χαρακτήρα. Οι γενετικές βλάβες που έχουν περιγραφεί εξαρτώνται από την καταγωγή των ασθενών, καθώς υπάρχουν γεωγραφικές διαφορές στις παρατηρούμενες μεταλλάξεις. Οι περισσότερες μεταλλάξεις έχουν περιγραφεί στην περιοχή της Μεσογείου, στη Μέση Ανατολή, την Ινδία και στο Εκουαδόρ. Μια περίπτωση κοριτσιού με συνδρόμου Laron έχει δημοσιευθεί στην Ελλάδα ( Γαλλή-Τσινοπούλου 2003). Η καταγωγή των παππούδων του παιδιού ήταν από την Μέση Ανατολή και η μητέρα του ήταν ετεροζυγώτης για την μετάλλαξη στο εξώνιο 4 που έφερε και το παιδί, χωρίς όμως αυτή να έχει οποιοδήποτε σύμπτωμα της νόσου. Συνήθως υπάρχει ιστορικό αιμομικτικών γάμων στις οικογένειες που έχουν πάσχοντα παιδιά. Έχουν περιγραφεί πάνω από 60 μεταλλάξεις (ομόζυγες και συνδυασμένες ετερόζυγες) και μόνο 3 απαλλείψεις. Οι περισσότερες (80%) των μεταλλάξεων του GHR αφορούν το εξωκυττάριο τμήμα του.
Οι μεταλλάξεις μπορούν να διακριθούν σε 3 τύπους ανάλογα με το αν αφορούν
α) στην έκφραση του γονιδίου GHR (τέτοια είναι μία σημειακή μετάλλαξη που ενεργοποιεί ένα ψευδοεξώνιο προσθέτοντας 108 νουκλεοτίδια μεταξύ του εξωνίου 6 και 7 του GHR, με αποτέλεσμα να επηρεάζει τον διμερισμό και τη μετάδοση του σήματος της GH)
β) στην ενεργοποίηση του GHR ( όπως η αντικατάσταση ιστιδίνης με ασπαργανικό οξύ στη θέση 152 του GHR) και
γ) στη μετάδοση του σήματος (παράδειγμα η μετάλλαξη σε τμήμα του εξωνίου 3 η οποία προκαλεί βράχυνση του κυτταροπλασματικού τμήματος του GHR, το οποίο με τον τρόπο αυτό στερείται λειτουργικότητας και αθροίζεται κατά μήκος της κυτταρικής μεμβράνης).
Ετερόζυγες γενετικές βλάβες του γονιδίου GHR είναι επίσης πιθανό να είναι υπεύθυνες για κοντό ανάστημα. Σε παιδιά με ύψος <-2 SDS, χαμηλά επίπεδα GHBP και IGF-1, έχουν βρεθεί σε ποσοστό περίπου 5-30% βλάβες στον GHR (Mullis 2011, Sanchez 1998), αν και άλλοι ερευνητές δεν έχουν διαπιστώσει επίδραση των ετερόζυγων μεταλλάξεων στο τελικό ανάστημα.
Τέλος, πολυμορφισμοί του γονιδίου GHR δεν φαίνεται να αποτελούν αιτία κλινικών διαταραχών. Ως πολυμορφισμός ορίζεται μια μεταβολή στην αλληλουχία του DNA που παρατηρείται σε τουλάχιστον 1% του πληθυσμού. Πολυμορφισμοί ενός μόνο νουκλεοτιδίου του γονιδίου GHR έχουν περιγραφεί στα εξώνια 6 και 10. Ο πολυμορφισμός του εξωνίου 3 του γονιδίου GHR είναι πιο σύνθετος και αφορά στην κωδικοποίηση μιας αλληλουχίας 22 αμινοξέων στο εξωκυττάριο τμήμα του GHR. Ασθενείς με έλλειψη GH ή άλλα αίτια χαμηλού αναστήματος που έχουν τουλάχιστον ένα τέτοιο αλλήλιο, έχει βρεθεί ότι έχουν καλύτερη απάντηση κατά τον πρώτο χρόνο θεραπείας με GH.
Θεραπεία. Η θεραπεία των ασθενών με σύνδρομο αντίστασης στην GH περιλαμβάνει θεραπεία με ανασυνδυασμένο IGF1. Η θεραπεία αυτή μπορεί να είναι αποτελεσματική σε μεταλλάξεις του GHR ή του σηματοδοτικού μονοπατιού ή του γονιδίου IGF-1. Xoρηγείται υποδορίως δύο φορές την ημέρα πριν ή εντός 20 λεπτών από το γεύμα σε δόση 60-120 μgr/kg. Η ταχύτυτα αύξησης του ύψους των παιδιών κατά τη θεραπεία με IGF1 είναι γενικά πιο μεγάλη κατά τον πρώτο χρόνο και μειώνεται κατά τα επόμενα χρόνια, αλλά παραμένει υψηλότερη από την ταχύτητα πριν την έναρξη της θεραπείας.
Κύρια παρενέργεια της θεραπείας είναι η υπογλυκαιμία, σπανιότερες η υπερτροφία αδενοειδών εκβλαστήσεων και η ενδοκράνια υπέρταση.Οι παρενέργειες αυτές μειώνονται με την πάροδο του χρόνου ή την παροδική διακοπή και επανέναρξη της αγωγής. Η ανταπόκριση στην θεραπεία με IGF1 είναι αποτελεσματική σε ασθενείς με σοβαρή πωτοπαθή έλλειψη IGF1 ή αυτούς που ανέπτυξαν αντισώματα κατά την διάρκεια της θεραπείας με GH, υπολείπεται όμως ελαφρά συγκρινόμενη με την ανταπόκριση ασθενών με έλλειψη GH στην αγωγή με ανασυνδυασμένη GH.
Οι μεταβολικές δράσεις της GH είναι εμφανείς σε περιπτώσεις ασθενών με έλλειψη GΗ. Αυτοί παρουσιάζουν κυρίως κοιλιακή παχυσαρκία, αύξηση του δείκτη μάζας σώματος και ελαττωμένη σύσταση σώματος, αύξηση των λιπιδίων, αυξημένο καρδιαγγειακό κίνδυνο και αύξηση προφλεγμονωδών κυτοκινών, παραγόντων πήξεως και ομοκυστείνης ήδη από την εφηβική ηλικία. Η λιπολυτική δράση της GH βελτιώνει τις διαταραχές των λιπιδίων και αναστρέφει τις βλάβες των αγγείων και τον κίνδυνο αθηρωματικής νόσου.
Ο IGF1 επιδρά άμεσα στην πρόσληψη της γλυκόζης από τους ιστούς και καταστέλλει την παραγωγή γλυκόζης από το ήπαρ. Σε ασθενείς με έλλειψη GH οι οποίοι παρουσιάζουν σχετική αντίσταση στην ινσουλίνη, η θεραπεία με IGF1, λόγω της ευαισθητοποιητικής δράσης στην ινσουλίνη και της μείωσης των επιπέδων γλυκόζης αίματος,μπορεί να είναι ευεργετική σε καταστάσεις π.χ διαβήτη. Επομένως ο συνδυασμός GH-IGF1 έχει αρκετά μεταβολικά πλεονεκτήματα (ελάττωση υπογλυκαιμιών από την χορήγηση IGF1, βελτίωση της ευαισθησίας στην ινσουλίνη που επηρεάζεται από την χορήγηση GH, προστιθέμενη αναβολική δράση) και βρίσκεται σε στάδιο κλινικών μελετών, τα αποτελέσματα των οποίων αναμένονται σύντομα.
3.2. Διαταραχές της μεταγωγής του σήματος της GH (μεταλλάξεις μετά τον υποδοχέα (post-GH receptor)
Όταν η GH συνδέεται με τον υποδοχέα της, ο οποίος βρίσκεται στην επιφάνεια του κυττάρου και τον διεγείρει, αρχίζει η λειτουργία του ενδοκυττάριου σηματοδοτικού καταρράκτη. Αρχικά ο υποδοχέας διμερίζεται και τα επόμενα στάδια περιλαμβάνουν αντιδράσεις φωσφορυλίωσης του JAK2 (Janus family tyrosine kinase 2) και εν συνεχεία ενεργοποίηση διαφόρων μονοπατιών, μεταξύ των οποίων και του STAT5b (signal transducer and activator of transcription).
Υπάρχουν 4 STAT πρωτεϊνες (STAT1,3,5a και 5b), οι οποίες ενεργοποιούνται από το σύστημα GH-GHR.Το σηματοδοτικό μονοπάτι του STAT5 έχει αποδειχθεί τόσο σε πειραματικά μοντέλλα τρωκτικών όσο και στον άνθρωπο ότι είναι το σημαντικότερο για την ρύθμιση της παραγωγής του IGF1.
Η πρωτεΐνη STAT5 μετακινείται και εισέρχεται στον πυρήνα για να συνδεθεί με την περιοχή εκκίνησης των IGF1, IGFBP3 και ALS και να αρχίσει η μεταγραφή των γονιδίων.
3.2.1. Διαταραχές του STAT 5
Η πρώτη περιγραφή μοριακής διαταραχής της μεταγωγής του σήματος μετά τον υποδοχέα της GH δημοσιεύθηκε το 2003 ( Kofoed 2003) και αφορούσε ομόζυγο μετάλλαξη του γονιδίου STAT5b.
Έκτοτε, έχουν δημοσιευθεί 6 ασθενείς με μεταλλάξεις αυτού του γονιδίου, οι οποίοι έχουν παρόμοια κλινικά και αυξολογικά χαρακτηριστικά με τους ασθενείς με αντίσταση στην αυξητική ορμόνη (GHIS), δηλαδή σοβαρή καθυστέρηση της αύξησης μετά την γέννηση, προέχον μέτωπο, ελαφρά υποπλασία του μέσου τριτημορίου του προσώπου και φυσιολογική περίμετρο κεφαλής. (Woods 2007, Bernasconi 2006, Hwa 2007, Vidarsdottir 2006).
Τα εργαστηριακά ευρήματα περιλαμβάνουν πολύ χαμηλές IGF1 (7-38 ng/ml) , IGFBP3 (180-874 ng/ml) και ALS με φυσιολογική GHBP λόγω της φυσιολογικής λειτουργίας του GHR.
Επιπλέον, ορισμένοι ασθενείς εμφανίζουν διαταραχές της ανοσίας των Τ λεμφοκυττάρων, λόγω της συμμετοχής του STAT 5b στην μεταγωγή του σήματος μέσω υποδοχέων των κυτοκινών. Οι κλινικές εκδηλώσεις περιλαμβάνουν λοιμώξεις του αναπνευστικού, διάμεσο πνευμονία ή σπανιότερα έρπητα ζωστήρα, χρόνιο έκζεμα και αιμορραγική μορφή ανεμευλογιάς. Επομένως θα πρέπει να διερευνάται η πιθανότητα διαταραχής του STAT5 σε παιδιά που παρουσιάζουν χρόνιες ή ανεξήγητες πνευμονοπάθειες σε συνδυασμό με σημαντική καθυστέρηση της αύξησης. Οι μισοί από τους ασθενείς παρουσιάζουν ήπια υπερπρολακτιναιμία, η οποία πιθανόν να οφείλεται επίσης στη διαταραχή της μετάδοσης του ενδοκυτταρίου σήματος της προλακτίνης.
3.2.2. Διαταραχές του STAT 3
Σε 4 ασθενείς με ISS αναφέρθηκε διαταραχή της ενεργοποίησης του STAT 3 και καλή ανταπόκριση στη χορήγηση GΗ με αποτέλεσμα άνοδο των επιπέδων του IGF1 και επιτάχυνση της αύξησης. (Rojas-Gil 2006 ). Επειδή όμως δεν ανευρέθη μετάλλαξη του γονιδίου, δεν μπορεί να στοιχειοθετηθεί με βεβαιότητα η διάγνωση (Rosenfeld 2007).
3.2.3. Άλλες διαταραχές
Μεταλλάξεις της φωσφατάσης της τυροσίνης Shp 2 ανιχνεύθηκαν σε ασθενείς με σύνδρομο Noonan,το οποίο χαρακτηρίζεται από χαμηλό ανάστημα, δυσμορφικά χαρακτηριστικά, καρδιοπάθεια και αυξημένο κίνδυνο λευχαιμίας.
3.3. Διαταραχές του IGF1 και του υποδοχέα του IGFR
Οι διαταραχές της αύξησης μπορεί επίσης να οφείλονται σε μεταλλάξεις γονιδίων που εδράζονται σε κατώτερα επίπεδα στον άξονα GH-IGF1: στο γονίδιο του IGF1 και στο γονίδιο του IGF1R.
Η πρώτη ανακοίνωση ασθενούς στον οποίο ανιχνεύθηκε μετάλλαξη του γονιδίου του IGF1 έγινε το 1996 (Woods 1996) και αφορούσε μια μεγάλη ομόζυγο απάλειψη του γονιδίου.
Έκτοτε έχουν παρατηρηθεί σημειακές μεταλλάξεις του γονιδίου IGF1 σε 4 ακόμα ασθενείς με σοβαρή ενδομήτρια καθυστέρηση της αύξησης, χαμηλό βάρος γέννησης (SGA) και καθυστέρηση αύξησης μετά την γέννηση, παρόμοια με τους ασθενείς με GHI. Οι ασθενείς με μεταλλάξεις του γονιδίου του IGF1 παρουσιάζουν μερικά τυπικά χαρακτηριστικά, που διαφέρουν από τους ασθενείς με GHI.
Εμφανίζουν χαμηλό βάρος γέννησης (ενδομήτρια καθυστέρηση), πιθανόν λόγω της μη εξαρτώμενης από την GH δράσης του IGF κατά την ενδομήτριο ζωή. Επίσης εμφανίζουν πνευματική καθυστέρηση, νευροαισθητηριακή κώφωση και ιδιόμορφο προσωπείο λόγω της έντονης μικροκεφαλίας, η οποία οφείλεται σε αντίστοιχη ανεξάρτητη της GH δράση του IGF1 στον αναπτυσσόμενο εγκέφαλο (Netchine 2009).
Η πρώτη ανακοίνωση μετάλλαξης του IGFR έγινε το 2003 σε έναν ασθενή με σοβαρού βαθμού ενδομήτρια καθυστέρηση της αύξησης , η οποία παρέμενε και μετά την γέννηση (Abbuzzahab 2003).
Έκτοτε έχουν δημοσιευθεί 8 ασθενείς με μετάλλαξη του IGFR. Ενδιαφέρον παρουσιάζει η περίπτωση 2 ασθενών με σημειακές μεταλλάξεις του IGFR, και φυσιολογικά ή αυξημένα επίπεδα IGF1. Πιθανόν στους ασθενείς αυτούς να παράγεται ένα μόριο IGF1, το οποίο ανιχνεύεται στον ορό, αλλά είναι βιολογικά ανενεργό. Στους ασθενείς με μεταλλάξεις του IGFR, η GH είναι φυσιολογική ή αυξημένη λόγω αντίστασης, αλλά τα επίπεδα IGFBP3, ALS και GHBP είναι φυσιολογικά, λόγω της φυσιολογικής λειτουργίας του υποδοχέα της GH.
Επίσης έχουν περιγραφεί περιπτώσεις ασθενών στην ίδια οικογένεια με ετερόζυγες μεταλλάξεις του IGFR και με ήπια καθυστέρηση της αύξησης (Walenkamp 2005) καθώς και ετερόζυγες μεταλλάξεις του IGFR σε ασθενείς με απάλειψη του 15q (Walenkamp 2013).
3.4. Διαταραχή του ALS
Το ALS αποτελεί τμήμα του τριτογενούς συμπλέγματος, το οποίο σταθεροποιεί τον IGF1 στην κυκλοφορία. Επειδή ο IGF1 αποσπάται από το σύμπλεγμα πριν εγκαταλείψει την κυκλοφορία και επειδή συντίθεται και τοπικά στους ιστούς, το σύμπλεγμα δεν επηρεάζει την τοπική (αυτοκρινή/παρακρινή δράση) του IGF1.
Η πρώτη περίπτωση έλλειψης ALS στον άνθρωπο δημοσιεύθηκε το 2004 (Domene 2004). Έκτοτε έχουν περιγραφεί τουλάχιστον 17 ασθενείς με ομόζυγες ή σύνθετες ετερόζυγες μεταλλάξεις του γονιδίου του ALS και μη ανιχνεύσιμα επίπεδα ALS (Domene 2007, Fofanova 2009).
Όλοι οι ασθενείς με μεταλλάξεις του γονιδίου του ALS παρουσιάζουν σημαντική ελάττωση των IGF1 και IGFBP3, συνήθως αυξημένη GH και φυσιολογική GHBP, η οποία οφείλεται σε φυσιολογική λειτουργία του υποδοχέα της GH. Παρά την σοβαρή έλλειψη του IGF1, οι ασθενείς με μεταλλάξεις του γονιδίου του ALS παρουσιάζουν μικρότερη καθυστέρηση αύξησης σε σύγκριση με ασθενείς που έχουν αντίστοιχα χαμηλές τιμές IGF1 οφειλόμενες σε άλλα γενετικά αίτια πρωτοπαθούς έλλειψης IGF.
Μάλιστα μερικοί από αυτούς έχουν ύψος κυμαινόμενο μέσα στα όρια του φυσιολογικού (SDS ύψους -0,5 έως -4,2), πιθανόν λόγω αυξημένης αυτοκρινούς /παρακρινούς παραγωγής IGF1. Μερικοί ασθενείς έχουν επίσης αντίσταση στην ινσουλίνη,οστεοπενία και καθυστέρηση ήβης (Hess, 2013).
Πάντως το κλινικό σύνδρομο της ανεπάρκειας του γονιδίου ALS δεν έχει αποσαφηνισθεί πλήρως και η κατάλληλη θεραπεία θα καθοριστεί από τα αποτελέσματα των μελλοντικών ερευνών.
4. Διερεύνηση ασθενών με διαταραχές του άξονα GH-IGF1 (δευτεροπαθής έλλειψη IGF1)
4.1. Οντογένεση υπόφυσης
H ανάπτυξη της υπόφυσης έχει μελετηθεί ιδιαίτερα στους μύς και θεωρείται ότι είναι παρόμοια σε όλα τα σπονδυλωτά, αλλά στον άνθρωπο ο ακριβής μηχανισμός ανάπτυξης της υπόφυσης δεν είναι πλήρως γνωστός. Η υπόφυση αναπτύσσεται από δύο διαφορετικές πηγές στο έμβρυο: o πρόσθιος και ο διάμεσος λοβός προέρχονται από το στοματικό εξώδερμα, ενώ ο οπίσθιος λοβός και ο μίσχος από ένα κοιλιακό εκκόλπωμα στο έδαφος του διεγκεφάλου (προερχομένου από το νευρικό εξώδερμα), το οποίο προεκβάλλει προς τα κάτω (Sadler 2006). Κατά την 3η εβδομάδα της εμβρυικής ανάπτυξης σχηματίζεται ένα εκκόλπωμα στην οροφή της αρχέγονης στοματικής κοιλότητας, το οποίο θα δώσει γένεση στον θύλακα του Rathke. Το εκκόλπωμα αυτό χάνει σταδιακά την σύνδεσή του με την στοματική κοιλότητα μέχρι το τέλος του 2ου μήνα της ανάπτυξης και επιμηκύνεται ραχιαία προς την αναπτυσσόμενη χοανοειδή εξοχή του διεγκεφάλου, με την οποία έρχεται σε επαφή. Τα κύτταρα της πρόσθιας επιφάνειας του θύλακα του Rathke πολλαπλασιάζονται γρήγορα και δίνουν γένεση στον πρόσθιο λοβό της υπόφυσης, εξαλείφεται σταδιακά ο αυλός και μένει μια μικρή υπολειμματική σχισμή, ενώ μια μικρή χοανοειδής επέκταση του λοβού προεκτείνεται προς τον μίσχο και τον περιβάλλει (Sadler 2006). Ο οπίσθιος λοβός της υπόφυσης αποτελείται κυρίως από νευρογλοιακά κύτταρα και νευρικές ίνες από την περιοχή του υποθαλάμου.
H οντογένεση της υπόφυσης, τόσο στα αρχικά στάδια, όσο και κατά την διαφοροποίηση των κυτταρικών σειρών που την αποτελούν, καθορίζεται από την έκφραση και την αλληλεπίδραση μιας σειράς σηματοδοτικών μορίων και μεταγραφικών παράγοντων. Ορισμένοι από τους παράγοντες που συμβάλλουν στην οργανογένεση εκφράζονται στον αναπτυσσόμενο θύλακο του Rathke, ενώ άλλοι στην περιοχή του κοιλιακού διεγκεφάλου και του αναπτυσσόμενου νευρικού συστήματος. Η οργανογένεση της υπόφυσης είναι το αποτέλεσμα μιας συντονισμένης χρονικά διαδικασίας, κατά την οποία η διαδοχική καταστολή και η ενεργοποίηση των γονιδίων- στόχων επιτρέπει να συνεχιστεί η κανονική ανάπτυξη.
Η έλλειψη GH είτε μεμονωμένη (isolated growth hormone deficiency, IGHD) είτε σε συνδυασμό με έλλειψη άλλων υποφυσιακών ορμονών (combined pituitary hormone deficiency, CPHD) είναι ένα σπάνιο νόσημα. Η επίπτωση αναφέρεται από 1:4000 έως 1:10 000 γεννήσεις, όπου η πλειοψηφία των περιπτώσεων είναι ιδιοπαθείς και μόνο 5–30% είναι οικογενείς (Dattani 2005). Tα τελευταία χρόνια η πρόοδος της μοριακής βιολογίας οδήγησε στην καλύτερη κατανόηση του νοσήματος. Γενετική αιτία ανευρίσκεται σε 10% περίπου των περιπτώσεων ανεπάρκειας GH. Στο παρακάτω κείμενο αναφέρονται εκείνα τα σηματοδοτικά μόρια και οι μεταγραφικοί παράγοντες που έχει αποδειχθεί ότι οι μεταλλάξεις τους στον άνθρωπο οδηγούν σε έλλειψη GH, είτε μεμονωμένη είτε σε συνδυασμό με πολλαπλή υποφυσιακή ανεπάρκεια, με επακόλουθο καθυστέρηση της αύξησης (Dattani 2005, Kelberman 2009, Pfäffle 2011, Cohen 2012). Οι παράγοντες αυτοί ταξινομούνται στις εξής κατηγορίες: 1. σε παράγοντες που έχουν σχέση με την ανάπτυξη της αρχέγονης υπόφυσης και άλλων περιοχών του εγκεφάλου (HESX1, LHX3 και LHX4, OTX2, PITX2, SOX3), 2. σε παράγοντες που έχουν σχέση κυρίως με την ανάπτυξη του διεγκεφάλου (GLI2) 3. σε παράγοντες που έχουν αποκλειστικά σχέση με την διαφοροποίηση της υπόφυσης (PROP1 και POU1F1) και 4. σε παράγοντες που προκαλούν οικογενή μεμονωμένη έλλειψη GH (IGHD τύπος IA, τύπος IB, τύπος ΙΙ, τύπος ΙΙΙ).
4.2. Παράγοντες που έχουν σχέση με την ανάπτυξη της αρχέγονης υπόφυσης
4.2.1. HESX1
Το γονίδιο HESX1 (ονομάζεται επίσης Rpx: Rathke’s pouch homeobox) χαρτογραφείται στη θέση 3p21.1–21·2 (ΟΜΙΜ 601802) και κωδικοποιεί ένα μεταγραφικό καταστολέα με άγνωστους ακόμη στόχους. Εκφράζεται στα πολύ πρώιμα στάδια της ανάπτυξης της αρχέγονης υπόφυσης και του προσθίου εγκεφάλου και πιθανόν να συμβάλλει σημαντικά στην αρχική ανάπτυξη του αδένα (Kelberman 2009, Cohen 2012). Το γονίδιο παύει να εκφράζεται όταν αρχίζει η διαφοροποίηση των κυττάρων της υπόφυσης, η δε εξάλειψη του είναι σημαντική για την ενεργοποίηση άλλων γονιδίων, όπως του PROP1 (Dattani 2005).
Mεταλλάξεις του γονιδίου αυτού στον άνθρωπο έχουν παρατηρηθεί σε άτομα με πολλαπλή υποφυσιακή ανεπάρκεια ή/και διαφραγματο-οπτική δυσπλασία (septo-optic dysplasia: SOD), που χαρακτηρίζεται από υποπλασία (ετερόπλευρη ή αμφοτερόπλευρη) του οπτικού νεύρου και της υπόφυσης ή/και ανωμαλίες των δομών της μέσης γραμμής του εγκεφάλου, όπως απλασία του μεσολοβίου και του διαφανούς διαφράγματος (Cohen 2012). Ο φαινότυπος του νοσήματος ποικίλλει και δεν υπάρχει σαφής συσχέτιση γονότυπου-φαινότυπου. Tο συχνότερο σύμπτωμα είναι η καθυστερημένη αύξηση λόγω ανεπαρκούς έκκρισης GH, ακολουθούν η έλλειψη TSH και ACTH και σπανιότερα συνυπάρχουν ανεπάρκεια γοναδοτροπινών και άποιος διαβήτης (Dattani 2005). Οι ενδοκρινικές διαταραχές εξελίσσονται και επιδεινώνονται με την πάροδο του χρόνου. Μπορεί να συνυπάρχουν νευρολογικές διαταραχές.
Μεταλλάξεις του γονιδίου HESX1 έχουν παρατηρηθεί τόσο σε ετεροζυγώτες με το σύνδρομο όσο και σε ομοζυγώτες, οπότε το νόσημα μπορεί να κληρονομείται είτε με τον υπολειπόμενο χαρακτήρα είτε με επικρατούντα χαρακτήρα και ατελή διεισδυτικότητα (Dattani 2005, Cohen 2012). Στους ετεροζυγώτες ο φαινότυπος είναι ηπιότερος. Η ακριβής αιτιολογία όμως παραμένει άγνωστη και πιθανόν να είναι πολυπαραγοντική. Περιβαλλοντικοί παράγοντες, όπως ιογενείς λοιμώξεις, ίσως να συμβάλλουν στην εμφάνιση του νοσήματος (Dattani 2005).
4.2.2. LHX3 και LHX4
Tα γονίδια LHX3 και LHX4 κωδικοποιούν μεταγραφικούς παράγοντες που ανήκουν στην ομάδα των LIM πρωτεϊνών και σχετίζονται με την ανάπτυξη της υπόφυσης. H ομοιοεπικράτεια LIM (homeodomain LIM) περιέχει δύο μοναδικές επαναλαμβανόμενες αλληλουχίες κυστεΐνης/ιστιδίνης, δεν έχει την ικανότητα να προσδένεται στο DNA, έχει όμως μεταγραφική δραστηριότητα και εκφράζεται στον αναπτυσσόμενο θύλακο του Rathke (Cohen 2012).
Ο παράγων LHX3 εκφράζεται στον πρόσθιο και διάμεσο λοβό της υπόφυσης και σε άλλα σημεία του αναπτυσσόμενου νευρικού συστήματος (Cohen 2012). Είναι ενδιαφέρον ότι η έκφρασή του στην υπόφυση διατηρείται κατά την ενήλικο ζωή, γεγονός που υποδηλώνει ότι ίσως συμβάλλει στην διατήρηση της λειτουργίας ενός ή περισσοτέρων κυτταρικών τύπων του πρόσθιου λοβού της υπόφυσης (Cohen 2012). Στον άνθρωπο ομόζυγες μεταλλάξεις που προκαλούν απώλεια λειτουργικότητας του παράγοντα LHX3 παρατηρήθηκαν σε ασθενείς με υποφυσιακή ανεπάρκεια και καθυστέρηση της αύξησης σε συνδυασμό με ανατομικές ανωμαλίες της υπόφυσης στην πλειοψηφία ασθενών. Στις περισσότερες περιπτώσεις, αλλά όχι πάντα, διατηρείται η έκκριση της ACTH. Μπορεί να συνυπάρχουν σκελετικές ανωμαλίες, όπως κοντός αυχένας με μειωμένη ικανότητα περιστροφής, δυσμορφία προσώπου, υπερεκτασιμότης των αρθρώσεων και σπανιότερα ελάττωση ή απώλεια της ακοής (Kelberman 2009, Cohen 2012) .
Όπως ο παράγων LHX3, έτσι και ο LHX4 εκφράζεται σε διάφορα σημεία του αναπτυσσόμενου εγκεφάλου και του νωτιαίου μυελού και συμβάλλει στην διαφοροποίηση και την ανάπτυξη των κυττάρων της υπόφυσης (Cohen 2012). Σε αντίθεση με τον LHX3, η έκφρασή του περιορίζεται στον πρόσθιο λοβό της υπόφυσης και μόνον στα αρχικά στάδια της ανάπτυξης της υπόφυσης (Sheng 1997). Στον άνθρωπο ετερόζυγες μεταλλάξεις του παράγοντα LHX4 εχουν ανιχνευθεί σε πολύ λίγες περιπτώσεις ασθενών με ποικίλης βαρύτητας υποφυσιακή ανεπάρκεια και ανατομικές ανωμαλίες της υπόφυσης (Kelberman 2009, Cohen 2012). Όλοι οι ασθενείς που έχουν περιγραφεί είχαν ανεπάρκεια της GH και ορισμένοι ανεπάρκεια της TSH, ACTH και σπανιότερα των γοναδοτροπινών.
4.2.3. OTX2
Το γονίδιο ΟΤΧ2 εκφράζεται νωρίς κατά την περίοδο σχηματισμού του τρίστιβου εμβρύου και η έκφρασή του είναι απαραίτητη για την διαμόρφωση του προσθίου εγκεφάλου (Kelberman 2009). Μεταλλάξεις του γονιδίου ΟΤΧ2 στον άνθρωπο ανιχνεύθηκαν σε άτομα με διαταραχές στην ανάπτυξη των οφθαλμών (μικροφθαλμία, ανοφθαλμία) σε ορισμένες περιπτώσεις σε συνδυασμό με ποικίλου βαθμού υποφυσιακή ανεπάρκεια, καθυστέρηση στην ανάπτυξη, ανατομικές διαταραχές της υπόφυσης και διαταραχές στην ανάπτυξη των ώτων (Kelberman 2009).
4.2.4 PITX2
Το γονίδιο PITX2, ομόλογο του PITX1, εκφράζεται αρχικά στην αρχέγονη στοματική κοιλότητα του εξωδέρματος και στη συνέχεια στον αναπτυσσόμενο θύλακο του Rathke, αλλά και σε άλλα σημεία του κεντρικού νευρικού συστήματος και της κοιλιακής χώρας (Cohen 2012). Η έκφραση του μεταγραφικού παράγοντα PITX2 στην αριστερή πλευρά είναι υπεύθυνη για τη ασύμμετρη ανάπτυξη οργάνων, όπως ο σπλήνας και η ασύμμετρη λόβωση των πνευμόνων. Η έκφρασή του διατηρείται κατά την ενήλικο ζωή σε ορισμένα κύτταρα της υπόφυσης, κυρίως στα θυρεοτρόπα και γοναδοτρόπα κύτταρα, αλλά όχι στα κορτικοτρόπα (Kelberman 2009, Cohen 2012). Μεταλλάξεις του γονιδίου στον άνθρωπο έχουν παρατηρηθεί σε ασθενείς με το σύνδρομο Axenfeld–Rieger. Το σύνδρομο αυτό κληρονομείται με τον αυτοσωματικό επικρατούντα χαρακτήρα και χαρακτηρίζεται από ανωμαλίες στην διαμόρφωση των οφθαλμών, υποπλασία οδόντων, προέχοντα ομφαλό, νοητική υστέρηση και σε ορισμένες σπάνιες περιπτώσεις έχουν αναφερθεί δυσπλασία της υπόφυσης με ελαττωμένη έκκριση GH (Kelberman 2009). Ο ρόλος του γονιδίου αυτού στην ανάπτυξη της υπόφυσης και στην υποφυσιακή ανεπάρκεια παραμένει υπό διερεύνηση.
4.2.5. SOX3
To γονίδιο SOX3 εκφράζεται σε όλο το κεντρικό νευρικό σύστημα, ιδιαίτερα στην περιοχή του κοιλιακού διεγκεφάλου και η έκφρασή του είναι απαραίτητη για την ανάπτυξη του θύλακα του Rathke. Μεταλλάξεις του SOX3, το οποίο βρίσκεται στο Χ χρωμόσωμα, έχουν σαν συνέπεια υποφυσιακή δυσλειτουργία ποικίλου βαθμού, συμπεριλαμβανομένης και μεμονωμένης έλλειψης GH και σε ορισμένες περιπτώσεις συνυπάρχουν διαταραχές στην ανάπτυξη του κρανίου και του προσώπου και νοητική υστέρηση (Kelberman 2009, Cohen 2012). Όλοι οι ασθενείς που έχουν περιγραφεί έως τώρα είναι άρρενες, ενώ οι θήλεις ετεροζυγώτες είναι φορείς της νόσου.
4.3. Παράγοντες που έχουν σχέση με την ανάπτυξη του διεγκεφάλου
4.3.1. GLI2
Το γονίδιο Gli2 ανήκει στην ομάδα μεταγραφικών παραγόντων GLI, που αποτελούν το σηματοδοτικό μονοπάτι του Sonic Hedgehog (SHH). Το σηματοδοτικό μονοπάτι shh θεωρείται ότι ρυθμίζει διαδικασίες όπως τον κυτταρικό πολλαπλασιασμό, τη διαφοροποίηση και τον σχηματισμό των ιστών και είναι καθοριστικό για την εμβρυϊκή ανάπτυξη στον άνθρωπο. Oι σηματοδοτικοί παράγοντες SHH/GLI2 εκφράζονται στον κοιλιακό διεγκέφαλο και είναι καθοριστικοί για την ανάπτυξη του θυλάκου του Rathke και την ανάπτυξη των κυτταρικών σειρών της υπόφυσης.
Μεταλλάξεις των γονιδίων της ομάδας SHH συσχετίζονται με ολοπροσεγκεφαλία- μια ανωμαλία στην ανάπτυξη του εγκεφάλου που χαρακτηρίζεται από ατελή διαχωρισμό των εγκεφαλικών ημισφαιρίων με αποτέλεσμα πολυδακτυλία, κρανιοπροσωπικές ανωμαλίες ποικίλης βαρύτητας, όπως κυκλωπία ή ανοφθαλμία, μικροκεφαλία, σχηματισμό ενός μόνο ρουθουνιού με τυφλό άκρο, παρουσία μονήρους κεντρικού κοπτήρα και άλλες διαταραχές (Roessler 1996, Kelberman 2009). Μεταλλάξεις του γονιδίου GLI2 στον άνθρωπο είναι σπάνιες και συσχετίζονται με διαταραχές στην λειτουργία της υπόφυσης (συμπεριλαμβανόμενης και μεμονωμένης έλλειψης GH), έκτοπο οπίσθιο λοβό υπόφυσης, με ή χωρίς ολοπροσεγκεφαλία (Kelberman 2009, Franca 2010, Franca 1013).
4.4. Παράγοντες που έχουν σχέση αποκλειστικά με την διαφοροποίηση της υπόφυσης
4.4.1. PROP1
Το γονίδιο PROP1 (Prophet του PIT-1 γονιδίου, ΟΜΙΜ 601538) χαρτογραφείται στο χρωμόσωμα 5q, κωδικοποιεί ένα μεταγραφικό παράγοντα , του οποίου η έκφραση περιορίζεται στον πρόσθιο λοβό της υπόφυσης κατά την διάρκεια της εμβρυικής ανάπτυξης (Cohen 2012). Κατά την ανάπτυξη της υπόφυσης, το PROP1 λειτουργεί καταστέλλοντας το γονίδιο HESX1 και ενεργοποιώντας το POU1F1, πιθανόν μέσω των σηματοδοτικών παραγόντων Wnt/β-catenin (Pfäffle 2011).
Οι μεταλλάξεις του γονιδίου PROP1 στον άνθρωπο επηρεάζουν την υποφυσιακή λειτουργία προκαλώντας πολλαπλή υποφυσιακή ανεπάρκεια (CPHD). Οι μεταλλάξεις του PROP-1 αποτελούν την συχνότερη αιτία οικογενούς πολλαπλής υποφυσιακής ανεπάρκειας (περίπου 50%), ενώ ανιχνεύονται σπανιότερα μεταξύ των σποραδικών μεταλλάξεων (Kelberman 2009). Οι μεταλλάξεις κληρονομούνται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα και αφορούν κυρίως στην περιοχή προσδεσης στο DNA. Η πιο συχνή μετάλλαξη (50–72% των οικογενών μεταλλάξεων) είναι η απάλειψη 2 ζευγών βάσεων 2 δινουκλοεοτιδίων (GA ή AG) μεταξύ τριών επαναλαμβανόμενων GA αλληλουχιών εντός του εξονίου 2, με αποτέλεσμα την αλλαγή του πλαισίου ανάγνωσης και την εισαγωγή ενός κωδικονίου λήξης (S109X), που οδηγεί σε ελαττωματική πρωτεΐνη, διακόπτοντας την πρόσδεση στο DNA και την ενεργοποίηση της μεταγραφής. Λόγω του ότι δεν μπορεί να προσδιοριστεί ποιο από τα τρία ίδια δινουκλεοτίδια λείπει, η μετάλλαξη αναφέρεται ως 296delGA ή c.301_302delAG. Η μετάλλαξη αυτή και η μετάλλαξη c.150delA αποτελούν περίπου το 97% του συνόλου των γνωστών μεταλλάξεων στο PROP1 (Kelberman 2009).
Η τυπική εικόνα του νοσήματος χαρακτηρίζεται από έλλειψη της GH, PRL και TSH και με την πάροδο της ηλικίας στις περισσότερες περιπτώσεις εμφανίζεται ανεπάρκεια γοναδοτροπινών και ACTH (Romero 2011). Ο φαινότυπος του νοσήματος ποικίλλει ακόμα και μεταξύ ασθενών με την ίδια μετάλλαξη (Cohen 2012). Επίσης ποικίλλει ο χρόνος έναρξης και η έκταση της υποφυσιακής ανεπάρκειας, στα περισσότερα όμως άτομα παρατηρείται προοδευτική επιδείνωση της υποφυσιακής ανεπάρκειας (Pfäffle 2011). Είναι ενδιαφέρον ότι έχουν περιγραφεί περιπτώσεις ασθενών με μεμονωμένη έλλειψη GH (Pfäffle 2011). Η καθυστέρηση της ανάπτυξης εμφανίζεται συνήθως νωρίς στη ζωή, η κλινική υποψία συνήθως τίθεται μετά τον 1ο χρόνο της ζωής (Mody 2002). Παρότι οι περισσότεροι ασθενείς εμφανίζουν καθυστερημένη αύξηση, έχουν περιγραφεί ασθενείς με μερική έλλειψη GH και υπογοναδισμό, οι οποίοι έφτασαν ένα σχεδόν φυσιολογικό τελικό ανάστημα, χωρίς να δοθεί θεραπεία με αυξητική ορμόνη (Arroyo 2002, Reynaud 2005). H ανεπάρκεια της TSH συνήθως έπεται της ανεπάρκειας της GH. Η ανεπάρκεια των γοναδοτροπινών ποικίλλει . στους περισσότερους ασθενείς δεν αρχίζει η εφηβεία, ορισμένοι μπαίνουν στην εφηβεία, αλλά εμφανίζουν διακοπή ή καθυστέρηση στην ολοκλήρωση των σταδίων της ήβης και ανεπαρκή απάντηση της FSH και LH στην διέγερση με GnRH (Μοdy 2002). Στα αγόρια μπορεί να παρατηρηθεί μικρό πέος και κρυψορχία. Με την πάροδο της ηλικίας μπορεί να εμφανιστεί και ανεπάρκεια της ACTH. Στις περισσότερες περιπτώσεις στην μαγνητική τομογραφία το μέγεθος της υπόφυσης είναι μικρό ή φυσιολογικό. Σε ορισμένες περιπτώσεις παρατηρείται μια μάζα στην υπόφυση, η οποία όμως υποστρέφεται με την πάροδο του χρόνου και δεν χρειάζεται χειρουργική παρέμβαση (Dattani 2005).
4.4.2. POU1F1
To γονίδιο POU1F1 χαρτογραφείται στο χρωμόσωμα 3p11 και κωδικοποιεί τον μεταγραφικό παράγοντα POU1F1, γνωστό και ως pituitary-specific transcription factor 1 (PIT1) ή growth hormone factor 1 (GHF1). Ο POU1F1 είναι μέλος της οικογένειας των μεταγραφικών παραγόντων POU, υπεύθυνων για την ανάπτυξη στα θηλαστικά. Το POU είναι ένα ακρωνύμιο από τα 3 πρώτα μέλη του που προσδιορίστηκαν: PIT-1 και OCT-1 στα θηλαστικά και UNC-86 στον νηματώδη σκώληκα C.elegans (http://omim.org/entry/173110). H πρωτείνη POU1F1 χαρακτηρίζεται από δύο περιοχές πρόσδεσης στο DNA, την POU-ομοιοπεριοχή (POU-homeo) και την POU-ειδική περιοχή (POU-specific), οι οποίες είναι απαραίτητες για την υψηλής συγγένειας πρόσδεση στους υποκινητές των γονιδίων της GH και της PRL (Dattani 2005). Ο POU1F1 εκφράζεται στον αναπτυσσόμενο πρόσθιο λοβό της υπόφυσης κατά την εμβρυική ζωή και συνεχίζει να εκφράζεται και κατά την ενήλικο ζωή (Kelberman 2009). Παίζει σημαντικό ρόλο στην ανάπτυξη και διαφοροποίηση των σωματοτρόπων, λακτοτρόπων και θυρεοτρόπων κυττάρων της υπόφυσης και συμβάλλει στην μεταγραφική ρύθμιση των γονιδίων της GH-1, PRL και TSH- β (Dattani 2005, Cohen 2012).
Περισσότερες από 35 μεταλλάξεις έχουν έως τώρα (2013) περιγραφεί (Kelberman 2009, Cohen 2012, Inoue 2012). Οι περισσότερες κληρονομούνται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα και η πιο συχνή είναι η αντικατάσταση αμινοξέος R271W και αφορά την POU-ομοιοπεριοχή του γονιδίου (Kelberman 2009, Cohen 2012). Η πρωτεΐνη που παράγεται από αυτήν τη μετάλλαξη προσδένεται στο DNA αλλά αναστέλλει την μεταγραφή.
Οι ασθενείς με μεταλλάξεις στο γονίδιο POU1F1 εμφανίζουν πλήρη ανεπάρκεια της GH και της PRL, που εκδηλώνεται τα πρώτα χρόνια της ζωής, ενώ η ανεπάρκεια της TSH παρατηρείται σε μερικούς, αλλά όχι σε όλους τους ασθενείς (Pfäffle 2011). Η παραγωγή κορτικοτρόπου ορμόνης και γοναδοτροπινών παραμένει φυσιολογική.O φαινότυπος όμως μπορεί να ποικίλλει και έχουν αναφερθεί ασθενείς με φυσιολογικά επίπεδα PRL (Gat-Yablonski 2002), αλλά και ασθενείς με φυσιολογική TSH (Gat-Yablonski 2002, Turton 2005). Η μαγνητική τομογραφία της υπόφυσης στους ασθενείς με μεταλλάξεις του γονιδίου POU1F1 δείχνει φυσιολογική ή υποπλαστική υπόφυση.
4.5. Παράγοντες που προκαλούν οικογενή μεμονωμένη έλλειψη GH
H οικογενής μεμονωμένη έλλειψη GH είναι ένα σπάνιο νόσημα. Τέσσερεις τύποι έχουν περιγραφεί που χαρακτηρίζονται και ταξινομούνται ανάλογα με τον τρόπο που κληρονομούνται: οι τύποι IA και IB κληρονομούνται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα, ο τυπος II με τον αυτοσωματικό επικρατούντα χαρακτήρα και ο τύπος ΙΙΙ με τον φυλοσύνδετο υπολειπόμενο χαρακτήρα (X-linked) (Alatzoglou 2012). Παρότι η προαναφερθείσα ταξινόμηση δεν βασίζεται σε κλινικά χαρακτηριστικά, έχουν παρατηρηθεί σε ορισμένες περιπτώσεις συσχετίσεις γονότυπου-φαινότυπου που έχουν κλινική σημασία.
4.5.1. Tύπος ΙΑ
Κληρονομειται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα και μόνο λίγες περιπτώσεις έχουν περιγραφεί εως σήμερα σε ασθενείς από οικογένειες με ιστορικό αιμομιξίας (Dattani 2005, Alatzoglou 2009). Το νόσημα οφείλεται σε μεταλλάξεις εντός του γονιδίου της GH-1, κυρίως μεγάλης έκτασης απαλλείψεις, έχουν περιγραφεί όμως και μικροαπαλλείψεις που οδηγούν σε ελαττωματική πρωτεϊνη. Το νόσημα χαρακτηρίζεται από σημαντική καθυστέρηση της ανάπτυξης και πλήρη έλλειψη GH. Αρχικά υπάρχει ανταπόκριση στην θεραπεία με GH, αργότερα όμως αναπτύσσονται αντισώματα έναντι της GH με αποτέλεσμα χαμηλό τελικό ανάστημα. Ο φαινότυπος των ασθενών ποικίλλει. Μπορεί να συνυπάρχει το χαρακτηριστικό προσωπείο της έλλειψης GH με υποπλασία μέσου τριτημορίου του προσώπου, καθυστέρηση οδοντοφυΐας και προέχον μέτωπο (Dattani 2005).
4.5.2. Tύπος ΙΒ
Κληρονομείται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα και οφείλεται σε ομόζυγες μεταλλάξεις ματίσματος (splice mutations) εντός του γονιδίου της GH-1 ή σε μεταλλάξεις στον υποδοχέα της GHRH (GHRH-R) (Dattani 2005).Αποτελεί τον συχνότερο από τους 3 τύπους. Οι ασθενείς εμφανίζουν ηπιότερο φαινότυπο από τον τύπο ΙΑ, με χαμηλά αλλά ανιχνεύσιμα επίπεδα GH και καθυστερημένη αύξηση, όμως ανταποκρίνονται καλά στην θεραπεία με GH και δεν αναπτύσσουν αντισώματα .
4.5.3. Τύπος ΙΙ
Κληρονομείται με τον αυτοσωματικό επικρατούντα χαρακτήρα και οφείλεται σε μεταλλάξεις του γονιδίου της GH-1, συνήθως μεταλλάξεις που επηρεάζουν το μάτισμα του γονιδίου εντός των εξι πρώτων νουκλεοτιδίων του εσονίου 3 (IVS3) (Alatzoglou 2012). Οι ασθενείς έχουν χαμηλά, αλλά ανιχνεύσιμα επίπεδα GH, καθυστέρηση αύξησης ποικίλης βαρύτητας και σε ορισμένες περιπτώσεις συνυπάρχει υποπλασία της υπόφυσης. Ανταποκρίνονται καλά στην θεραπεία με GH και δεν αναπτύσσονται αντισώματα έναντι της GH. Είναι ενδιαφέρον ότι έχουν αναφερθεί περιπτώσεις όπου συνυπάρχει έλλειψη και άλλων υποφυσιακών ορμονών, όπως της TSH, ACTH, PRL και γοναδοτροπινών (Dattani 2005, Alatzoglou 2012).
4.5.4. Τύπος ΙΙΙ
Κληρονομείται με τον φυλοσύνδετο υπολειπόμενο χαρακτήρα και έχει περιγραφεί σε άρρενα άτομα, στα οποία συνυπάρχει εκτός από την έλλειψη GH και ανεπάρκεια γ-ανοσοσφαιρινών (Dattani 2005). Η γενετική βάση του νοσήματος παραμένει άγνωστη. Στους ασθενείς που φέρουν το νόσημα δεν έχουν παρατηρηθεί διαταραχές στο γονίδιο της GH1, ενώ πιθανόν να ευθύνονται μεταλλάξεις σε άλλα γονίδια, όπως στο γονίδιο btk (Alatzoglou 2012).
5. Συμπεράσματα
Η σημασία του άξονα GH-IGF1 είναι θεμελιώδης και βοηθά στην κατανόηση των διαταραχών που προκύπτουν όταν υπάρχει βλάβη σε οποιοδήποτε σημείο του άξονα. Το αποτέλεσμα θα είναι διαταραχή της αύξησης, η οποία μπορεί να παρατηρηθεί από την αρχή της εξωμήτριας ζωής και κατά την διάρκεια της παιδικής ηλικίας και να οδηγήσει σε χαμηλό ανάστημα στην ενήλικο ζωή.
Επομένως είναι πολύ σημαντικό να τεθεί η διάγνωση του ασθενούς σε όσο το δυνατόν μικρότερη ηλικία, ώστε να μπορεί να δοθεί η κατάλληλη θεραπεία. Υπάρχει μεγάλη πρόοδος στην γενετική διερεύνηση των διαταραχών του άξονα GH-IGF1, η οποία επιτρέπει την ταξινόμηση των ασθενειών με βάση όχι μόνον τα κλινικά και βιοχημικά χαρακτηριστικά, αλλά και την μοριακή βλάβη που υποκρύπτεται. Συγκεκριμένα, υπάρχει σημαντική πρόοδος στην κατανόηση της οντογένεσης της υπόφυσης και στην ταυτοποίηση των γονιδίων που συμβάλλουν στην ομαλή ανάπτυξή της. Επίσης έχουν ταυτοποιηθεί πολλά γονίδια τα οποία σχετίζονται με διαταραχές σε κατώτερα σημεία του άξονα GH-IGF1και οδηγούν σε πρωτοπαθή ανεπάρκεια IGF1, με αποτέλεσμα αντίσταση στη δράση της GH (GHIS). Ο προσδιορισμός της γενετικής διαταραχής μπορεί να βοηθήσει σημαντικά στην κλινική διάγνωση, την παρακολούθηση της πορείας του νοσήματος και την κατάλληλη θεραπεία. Επίσης, η γνώση του υπεύθυνου γονιδίου για εκδήλωση υποφυσιακής ανεπάρκειας καθορίζει την αναγκαιότητα τακτικής παρακολούθησης για πιθανές ορμονικές διαταραχές που θα εκδηλωθούν σταδιακά. Η ακριβής αιτιολογία της διαταραχής παραμένει εντούτοις ασαφής σε αρκετούς ασθενείς και αποτελεί πρόκληση για το μέλλον. Παράλληλα ερευνώνται νέες θεραπευτικές μέθοδοι, όπως η χορήγηση ανασυνδυασμένου IGF1 ή συνδυασμού GH-IGF1, ώστε να μεγιστοποιηθούν τα οφέλη από την θεραπεία των ασθενών αυτών.
Βιβλιογραφία
- Rosenfeld RG. Insulin-like growth factors and the basis of growth. N Engl J Med 2003, 349: 2184-2186
- Savage, M.O, Burren C,Rosenfeld R.(2010) The continuum of growth hormone-IGF 1 axis defects causing short stature diagnostic and therapeutic challenges. Clin Endocrinol 2010 72: 721-728
- Cohen P. Overview of the IGF system. Horm Res 2006, 65(suppl 1):3-8
- Cohen P. Controversy in clinical endocrinology: problems with reclassification of insulin-like growth factor I production and action disorders. J Clin Endocrinol Metab 2006, 91: 4235-4236
- Domene HM, Bengolea SV, Martinez AS et al. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. N Engl J Med 2004, 349: 2184-2186
- Lupu F, Terwilliger JD, Kaechong L et al. Roles of growth hormone and insulin-like growth factor-1 in mouse postnatal growth. Develop Biol 2001, 229: 141-162
- Woods KA, Camacho-Hubner C, Savage MO et al. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin –like growth factor-I gene. N Engl J Med 1996, 355: 1363-1367
- Walenkamp MJ, van der Kamp HJ, Pereira et al. A variable degree of intrauterine and post natal growth retardation in a family with a missense mutation in the insulin-like growth factor I receptor. J Clin Endocrinol Metab 2006, 91: 3062-3070
- Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clin Endocrinol (Oxf) 2005, 63(2): 121-30.
- Russo VC, Gluckmann P, Feldmann E et al. The insulin-like growth factor system and its pleiotrpic functions in brain. Endocrin Rev 2005, 26: 916-943
- Ranke M (2006). Defining insulin-like growth factor –I deficiency. Horm Res 2006, 65 (suppl 1): 9-14
- Woods KA, Dastot F, Preece MA et al. Phenotype-genotype relationships in growth hormone insensitivity syndrome. J Clin Endocrinol Metab 1997, 82: 3529-3535
- Wit JM, Clayton PE, Rogol AD et al. Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth hormone & IGF Res 2008, 18: 89-110
- Guevarra-Aguirre J, Rosenbloom A.L,Guevarra-Aguirre M et al. Effects of heterogygosity for the E 180 splice site mutation causing growth hormone receptor deficiency in Equador,on IGF1, IGFBP3 and stature. Growth hormone & IGF Res 2007, 17: 261-264
- Savage MO, Attie KM, David A et al. Endocrine assessment, molecular characterization and treatment of growth hormone insensitivity disorders. Nat Clin Pract Endocrinol Metab 2006, 72: 395-407
- Rosenfeld RG, Belgorosky A, Camacho-Hubner C et al. Defects in growth hormone receptor signaling. Trends Endocrinol Metab 2007, 18: 134-141
- Domene HM, Hwa V, Argente J et al. Human acid labile subunit:clinical, endocrine and metabolic consequences. Horm Res 2009, 72: 129-141
- Walenkamp MJ & Wit JM. Single gene mutations causing SGA. Best Pract Res Clin Endocrinol Metab 2008, 22: 433-446
- Rudman D, Kutner MH, Blackston RD, Cushman RA, Bain RP, Patterson JH. Children with normal-variant short stature : treatment with human growth hormone for six months. N Engl J Med 1981, 305: 123-131
- Bloom WF, Cotteril AM, Postel-Vinay MC, Ranke MB et al. Improvement of diagnostic criteria in growth hormone insensitivity syndrome: solutions and pitfalls .Pharmacia Study Group on Insulin-Like-Growth Factor I Treatment in Growth Hormone Insensitivity Syndromes. Acta Paediatr Suppl 1994; 399: 117-124
- Buckway CK, Guevarra-Aguirre J, Pratt KL et al.The IGF generation test revisited: a marker of GH sensitivity. J Clin Endocrinol Metab 2001, 86: 5176-5183
- Buckway CK, Selva KA, Pratt KL et al. Insulin-like growth factor binding-protein -3 generation as a measure of GH sensitivity. J Clin Endocrinol Metab 2002, 87: 4754-65
- Woods KA, Dastot F, Preece MA et al (1997) Phenotype-genotype relationships in growth hormone insensitivity syndrome. J Clin Endocrinol Metab 1997, 82: 3529-3535
- Backeljauw PF, Chernausek SD. The insulin-like growth factors and growth disorders of childhood. Endocrinol Metab Clin N Am 2012, 41: 265-282
- Walenkamp MJE, Wit JM. Genetic disorders in the growth hormone-insulin-like growth factor-I axis. Horm Res 2006, 66:221-230.
- Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003. J Clin Endocrinol Metab 2004, 89 (3): 1031-1044
- Galli-Tsinopoulou A, Nousia-Arvanitakis S, Tsinopoulos I et al. Laron syndrome. First report from Greece. Hormones 2003, 2 (2): 120-124
- Mullis P-E. Genetics of GHRH, GHRH receptor, GH-receptor: its impact on pharmacogenetics. Best Pract Res Clin Endocrinol Metab 2011, 25: 25-41
- Sanchez JE, Perera E, Baumbach L, Cleveland WW. Growth hormone receptor mutations in children with idiopathic short stature. J Clin Endocrinol Metab 1998, 83 (11): 4079-4083.
- Kofoed EM, Hwa V, Little B, et al Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med 2003, 349 (12): 1139-47
- Woods K. Genetic defects of the growth hormone-IGF axis associated with growth hormone insensitivity. Endocr Dev 2007, 11: 6-15
- Bernasconi A, Marino R, Ribas A et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics 2006, 118(5): 218-224
- Hwa V, Camacho-Hubner C, Little BM et al. Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon 13- intron 13 junction of the STAT5b gene. Horm Res 2007, 68 (5): 218-224
- Vidarsdottir S, Walenkamp MJ, Pereira AM, et al. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J Clin Endocrinol Metab 2006, 91 (9): 3482-3485
- Rojas-Gil AP, Ziros PG, Diaz L,et al (2006) Growth hormone/JAK-STAT axis signal-transduction defect. A novel treatable cause of growth failure. FEBS J 2006, 273:3454-3466
- Netchine I, Azzi S, Houang M et al. Partial primary deficiency of insulin-like growth factor (IGF1) activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab 2009, 94 (10): 3913-3921
- Abbuzzahab MJ, Schneider A, Goddart A et al. IGF1 receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med 2003, 349: 2211-2222.
- Walenkamp M.J, Karperien M, Pereira A.M et al. Homozygous and heterozygous expression of a novel insulin-like growth factor –I mutation. J Clin Endocrinol Metab 2005, 90 (5): 2855-2864
- Walenkamp MJ, Losekoot M, Wit JM. Molecular IGF-1 and IGF-1 receptor defects: from genetics to clinical management. Endocr Dev.2013, 24:128-37
- Domene HM, Scaglia PA, Lteif A et al. Phenotypic effects of null and haploinsufficiency of acid labile subunit in a family with two novel IGFALS gene mutations. J Clin Endocrinol Metab 2007, 92 (11): 4444-4450
- Fofanova-Gambetti OV, Hwa V, Kirsch S et al. Three novel IGFALS gene mutations resulting in total ALS and severe circulating IGF1/IGFBP3 deficiency in children of different ethnic origins. Horm Res 2009, 71 (2):100-110
- Hess O, Khayat M, Hwa V, et al. A novel mutation in IGFALS, c.380T>C (p.L127P), associated with short stature, delayed puberty, osteopenia and hyperinsulinaemia in two siblings: insights into the roles of insulin growth factor-1 (IGF1). Clin Endocrinol (Oxf). 2013, Mar 15. doi: 10.1111/cen.12200
- Sadler TW, 2006 Central Nervous system: In: Lippincott Williams Wilkins, (eds) Langman’s Medical Embryology 10th edition, Baltimore, Maryland, USA; pp, 285-316
- Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clin Endocrinol (Oxf). 2005, 63(2):121-130
- Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. 2009, 30(7):790-829
- Pfäffle R, Klammt J. Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab. 2011, 25(1):43-60.
- Cohen LE. Genetic disorders of the pituitary. Curr Opin Endocrinol Diabetes Obes. 2012, 19(1):33-39
- Sheng HZ, Moriyama K, Yamashita T, et al. Multistep control of pituitary organogenesis. Science. 1997, 278(5344):1809-1812
- Roessler E, Belloni E, Gaudenz K, et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996, 14(3):357-360
- França MM, Jorge AA, Carvalho LR, et al. Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J Clin Endocrinol Metab. 2010, 95(11):E384-91
- França MM, Jorge AA, Carvalho LR, et al. Relatively high frequency of non-synonymous GLI2 variants in patients with congenital hypopituitarism without holoprosencephaly. Clin Endocrinol (Oxf). 2013, 78(4):551-557
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. Mol Endocrinol. 2011 Jun 9;46(3):R93-R102
- Mody S, Brown MR, Parks JS. The spectrum of hypopituitarism caused by PROP1 mutations. Best Pract Res Clin Endocrinol Metab. 2002, 16(3):421-431
- Arroyo A, Pernasetti F, Vasilyev VV, Amato P, Yen SS, Mellon PL. A unique case of combined pituitary hormone deficiency caused by a PROP1 gene mutation (R120C) associated with normal height and absent puberty. Clin Endocrinol (Oxf). 2002, 57(2):283-291
- Reynaud R, Barlier A, Vallette-Kasic S et al. An uncommon phenotype with familial central hypogonadism caused by a novel PROP1 gene mutant truncated in the transactivation domain. Clin Endocrinol Metab. 2005, 90(8):4880-4887
- Inoue H, Mukai T, Sakamoto Y et al, Japan Growth Genome Consortium. Identification of a novel mutation in the exon 2 splice donor site of the POU1F1/PIT-1 gene in Japanese identical twins with mild combined pituitary hormone deficiency. Clin Endocrinol (Oxf) 2012, 76(1):78-87
- Gat-Yablonski G, Lazar L, Pertzelan A, Phillip M. A novel mutation in PIT-1: phenotypic variability in familial combined pituitary hormone deficiencies. J Pediatr Endocrinol Metab 2002, 15(3):325-330
- Turton JP, Reynaud R, Mehta A et al. Novel mutations within the POU1F1 gene associated with variable combined pituitary hormone deficiency. J Clin Endocrinol Metab. 2005, 90(8):4762-4770
- Alatzoglou KS, Dattani MT. Phenotype-genotype correlations in congenital isolated growth hormone deficiency (IGHD). Indian J Pediatr. 2012, 79(1):99-106
- Alatzoglou KS, Turton JP, Kelberman D et al. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. 2009, 94(9):3191-3199
Created: February 25, 2015
Last update: February 25, 2015