
Α. Δριμάλα, Ε. Μπίλλα, Ε. Βενάκη και Σ.Χ. Νικοπούλου
Ενδοκρινολόγοι
1. Εισαγωγή
Υπογοναδισμός είναι η έκπτωση της λειτουργίας της γονάδας, ανδρικής ή γυναικείας, λόγω βλάβης σε οποιοδήποτε σημείο του υποθαλαμο-υποφυσιακο-γοναδικού άξονα. Στην περίπτωση του άρρενος φύλου είναι: το κλινικό σύνδρομο που προκύπτει από την αδυναμία των όρχεων να παράγουν επαρκείς ποσότητες ανδρογόνων ή/και σπερματοζωαρίων λόγω βλάβης του υποθαλαμο-υποφυσιακο-ορχικού άξονα σε ένα ή περισσότερα επίπεδα1. Όταν η βλάβη αφορά τον υποθάλαμο ή/και την υπόφυση με αποτέλεσμα την ανεπαρκή παραγωγή, έκκριση ή/και δράση των γοναδοτροπινών FSH και LH ο υπογοναδισμός ονομάζεται υπογοναδοτροπικός και συνήθως οδηγεί σε ανεπάρκεια όλων των κυτταρικών πληθυσμών και σπανιότερα μεμονωμένα των κυττάρων Leydig ή Sertoli. Ο υπογοναδοτροπικός υπογοναδισμός χαρακτηρίζεται από χαμηλά επίπεδα γοναδοτροπινών, τεστοστερόνης, ινσουλινόμορφου παράγοντα 3 (INSL3) και διαταραχές της σπερματογένεσης. Τα αίτιά του μπορεί να συγγενή ή επίκτητα (πίνακας 1). Αξίζει να σημειωθεί ότι η πιο συχνή αιτία υπογοναδοτροπικού υπογοναδισμού (ΥΥ) είναι παροδική και ονομάζεται ιδιοσυστασιακή καθυστέρηση της ανάπτυξης και της ήβης (CDGP: Congenital Delay of Growth and Puberty)2,3,4.
Η κλινική εικόνα του υπογοναδισμού εξαρτάται από την ηλικία εμφάνισής του, τη βαρύτητα και διάρκεια της διαταραχής, τον πάσχοντα κυτταρικό πληθυσμό, την ευαισθησία των ιστών στόχων στα ανδρογόνα, τη γενικότερη κατάσταση υγείας και την προηγούμενη χρήση ανδρογόνων5. Στο σημείο αυτό πρέπει να τονισθεί ότι ο υπογοναδοτροπικός υπογοναδισμός δεν εκδηλώνεται ποτέ με αμφίβολα έξω γεννητικά όργανα, που είναι χαρακτηριστική εικόνα του υπογοναδισμού του πρώτου τριμήνου, καθώς η λειτουργία των κυττάρων Leydig και επομένως η παραγωγή τεστοστερόνης στην περίοδο αυτή είναι ανεξάρτητη από τη δράση των γοναδοτροπινών, αλλά βρίσκεται υπό τον έλεγχο της χοριακής γοναδοτροπίνης (hCG). Επομένως, ο υπογοναδοτροπικός υπογοναδισμός εμβρυϊκής έναρξης εμφανίζεται μετά το δεύτερο τρίμηνο και εκδηλώνεται με μικροφαλία, μικρούς όρχεις και κρυψορχία, ενώ όταν εμφανίζεται στην παιδική ή εφηβική ηλικία με απουσία ή αναστολή της ήβης6 .
Πίνακας 1. Αίτια υπογοναδοτροπικού υπογοναδισμού
| Συγγενή | Επίκτητα |
| Ολική ορχική ανεπάρκεια | Ολική ορχική ανεπάρκεια |
| Σ. Kallmann | Όγκοι υποθαλάμου – υπόφυσης |
| Ιδιοπαθής υπογοναδοτροπικός υπογοναδισμός | Τραύμα ή ακτινοβολία |
| Σ. Prader Willi | Αγγειακές βλάβες (ισχαιμία, αποπληξία της υπόφυσης) |
| Σ. Laurence-Moon-Biedl | Λοιμώξεις (φυματίωση, AIDS, σύφιλη) |
| ΥΥ και συγγενής υποπλασία επινεφριδίων | Υποθυρεοειδισμός |
| Πολλαπλή ανεπάρκεια υποφυσιακών ορμονών | Άλλες ενδοκρινοπάθειες και μεταβολικά νοσήματα (σ. Cushing, υπερπρολακτιναιμία, σακχαρώδης διαβήτης) |
| Δυσλειτουργία των κυττάρων Leydig | Διηθητικές νόσοι (αιμοχρωμάτωση, σαρκοείδωση, ιστιοκύττωση, κοκκιωματώδεις νόσοι, λεμφοκυτταρική υποφυσίτιδα |
| Μεταλλάξεις β-υπομονάδας LΗ | Χρόνια νοσήματα και κακή γενική υγεία |
| Μεταλλάξεις TAC, TACR 3 | Διαταραχές πρόσληψης τροφής (παχυσαρκία, νευρογενής ανορεξία, βουλιμία) |
| Δυσλειτουργία των κυττάρων Sertoli | Κατάχρηση αλκοόλ, οπιοειδών, κορτικοειδών |
| Μεταλλάξεις β-υπομονάδας FSH | Χρήση αναβολικών στεροειδών |
Στοιχεία φυσιολογίας
Ο γοναδικός άξονας ρυθμίζεται από ένα πλέγμα νευρώνων (hypothalamic pulse generator), που εντοπίζεται στο μεσο-βασικό υποθάλαμο, και ελέγχεται διεγερτικά από την πεπτιδική ορμόνη κισπεπτίνη (kisspeptin) η οποία παράγεται επίσης στον υποθάλαμο. Εκεί και στον τοξωτό πυρήνα του υποθαλάμου συντίθεται ένα δεκαπεπτίδιο, η εκλυτική των γοναδοτροπινών ορμόνη (Gonadotropin Releasing Hormone-GnRH), η οποία απελευθερώνεται κατά ώσεις, κάθε 60-90 λεπτά, και διεγείρει την επίσης κατά ώσεις απελευθέρωση στην κυκλοφορία των γοναδοτροπινών LH και FSH. Στη συνέχεια, οι γοναδοτροπίνες δρουν στον όρχι, η μεν LH διεγείροντας τα κύτταρα Leydig για την παραγωγή της τεστοστερόνης η δε FSH δρώντας στα κύτταρα Sertoli επάγει τη σπερματογένεση (σε συνέργεια με την ενδο-ορχική τεστοστερόνη αλλά και πλείστους παρακρινικούς παράγοντες).
Στους όρχεις παράγονται καθημερινά 3-10 mg τεστοστερόνης, ποσότητα που αντιστοιχεί σε επίπεδα ορού 10,4 – 34,7 nmol/L. Η δράση της ορμόνης στους ιστούς – στόχους γίνεται είτε μέσω του ίδιου του μορίου της (το οποίο συνδέεται με τους ανδρογονικούς υποδοχείς), είτε μέσω μετατροπής της στους δύο ενεργούς μεταβολίτες της, τη διυδροτεστοστερόνη (με αναγωγή) και την οιστραδιόλη (με αρωματοποίηση) με τελικό αποτέλεσμα τη διέγερση της σπερματογένεσης και της αρρενοποίησης. Ταυτόχρονα αναστέλλει παλίνδρομα την έκκριση της GnRH από τον υποθάλαμο τόσο με τη μορφή ανδρογόνων (τεστοστερόνη και διυδρoτεστοστερόνη) όσο και μεταβολιζόμενη σε οιστρογόνα τα οποία ασκούν αρνητική παλίνδρομη ρύθμιση δρώντας σε ειδικούς υποδοχείς στους νευρώνες του τοξωτού πυρήνα του υποθαλάμου οι οποίοι εκκρίνουν κισπεπτίνη 7,8,9,10.
Η GnRH pulse generator αποτελεί τον κύριο ρυθμιστή της έναρξης της ήβης. Η παραγωγή GnRH ξεκινά νωρίς στην εμβρυϊκή ζωή. Αυτό έχει ως αποτέλεσμα τα επίπεδα των γοναδοτροπινών να αλλάζουν σημαντικά κατά τη διάρκεια της εμβρυϊκής ανάπτυξης, της παιδικής ηλικίας, της εφηβείας και της ενηλίκου ζωής (σχήμα 1). Κατά τους πρώτους έξι μήνες της ζωής του βρέφους παρατηρείται έντονη γοναδική λειτουργία ως ανταπόκριση στη δράση των γοναδοτροπινών (mini- puberty). Μετά από αυτήν την περίοδο ακολουθεί πτώση των επιπέδων των γοναδοτροπινών μέχρι την έναρξη της ήβης, οπότε επανενεργοποιείται ο υποθαλαμο-υποφυσιακο-γοναδικός άξονας.

Σχήμα 1: Σχηματική παράσταση των γοναδοτροπινών και των παραγώμενων από τους όρχεις ορμονών κατά την ανάπτυξη του όρχεως (Rey RA, et al. Andrology. 2013;1:3-16).
2. Μεμονωμένος / Ιδιοπαθής υπογοναδοτροπικός υπογοναδισμός (Isolated / Idiopathic Hypogonadotropic Hypogonadism- ΙΥΥ) και Σύνδρομο Kallmann
Είναι σπάνια διαταραχή, με επίπτωση 1:10.000, που οφείλεται σε διαταραχή της έκκρισης ή της δράσης της GnRH με αποτέλεσμα την ανεπάρκεια των γοναδοτροπινών (FSH και LH) και κατά συνέπεια των στεροειδών του φύλου (τεστοστερόνη ή οιστραδιόλη). Η κλινική εικόνα καθορίζεται από το είδος και τη βαρύτητα της υποθαλαμικής διαταραχής. Ο ΥΥ με συνοδό διαταραχή της όσφρησης (ανοσμία/ υποσμία) αναφέρεται ως σύνδρομο Kallmann ενώ, ο μεμονωμένος ΥΥ με φυσιολογική όσφρηση (νορμο-οσμικός) ως ιδιοπαθής (ΙΥΥ) 8,11,12.
Αιτιοπαθογένεια – Γενετική
Ο ΙYY και το σύνδρομο Κallmann έχουν κοινή παθοφυσιολογική βάση, η οποία συνίσταται σε διαταραχή στην έκκριση ( από τον υποθάλαμο) ή σε διαταραχή της δράσης της GnRH στα γοναδοτρόπα κύτταρα της υπόφυσης, λόγω μετάλλαξης του υποδοχέα της.
Κατά τη διάρκεια της εμβρυϊκής ανάπτυξης οι πρόδρομοι νευρώνες της GnRH μεταναστεύουν από το οσφρητικό επιθήλιο στην τελική τους θέση στο μεσο-βασικό υποθάλαμο, όπου ολοκληρώνουν τη διαφοροποίησή τους και ξεκινούν την έκκριση της ορμόνης 13,14. Η σωστή ανάπτυξη και συντονισμένη λειτουργία των νευρώνων και των γοναδοτρόπων κυττάρων είναι θεμελιώδης για τη σωστή λειτουργία των γονάδων του εμβρύου και του νεογνού (mini- puberty). Μετά από μια περίοδο σιγής κατά τη βρεφική και παιδική ηλικία, ο άξονας υποθάλαμος-υπόφυση-γονάδες ενεργοποιείται ξανά κατά την εφηβεία και παραμένει ενεργός καθ΄ όλη τη διάρκεια της ενήλικης ζωής (παραγωγική περίοδος) 15.
Και οι δύο τύποι ΥΥ (νορμο-οσμικός ή ανοσμικός ΥΥ) μπορεί να κληρονομούνται με σωματικό επικρατούντα, με σωματικό υπολειπόμενο ή με φυλοσύνδετο τρόπο. Στην πλειοψηφία τους, όμως, πρόκειται για σποραδικές περιπτώσεις. Κατά την τελευταία δεκαετία έχει σημειωθεί σημαντική πρόοδος προς την κατεύθυνση της διερεύνησης της γενετικού υποστρώματος στον ΙΥΥ (Πίνακας 2). Μέχρι στιγμής έχουν αναγνωρισθεί περίπου 25 μεταλλάξεις υπεύθυνες για το 50% των ασθενών που πάσχουν από σύνδρομο Kallmann και ΙΥΥ 2,16,17,18,19. Επιπροσθέτως, έχει φανεί ότι οι ίδιες γενετικές μεταλλάξεις συσχετίζονται τόσο με το σύνδρομο Kallmann όσο και με τον νορμο-οσμικό ΙΥΥ υπονοώντας ότι δεν θα πρέπει πλέον να θεωρούνται διαφορετικές οντότητες8,20. Στην πραγματικότητα παρατηρείται σημαντική φαινοτυπική ετερογένεια σε οικογένειες με σύνδρομο Kallmann, στις οποίες κάποια μέλη εμφανίζονται με μεμονωμένη ανοσμία, άλλα με νορμο- οσμικό ΥΥ και άλλα με κλασσικό σύνδρομο Kallmann 11.
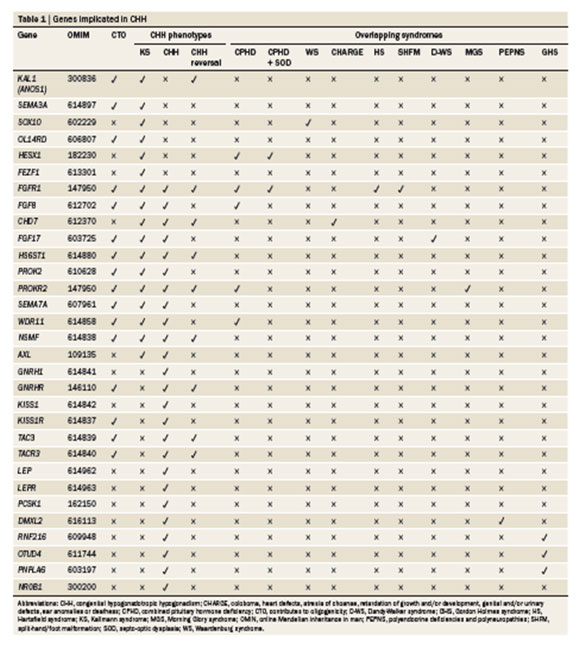
Πίνακας 2: Γονίδια υπεύθυνα για την αιτιοπαθογένεια του σ. Κallmann ή του ΙΥΥ (από Pitteloud, et al. Nat. Rev. Endocrinol 2015; 11:547-564).
Τα γονίδια που εμπλέκονται στην αιτιοπαθογένεια του σ. Kallmann και του ΙΥΥ είναι εκείνα που καθορίζουν την εμβρυϊκή διαφοροποίηση και τον πολλαπλασιασμό των GnRH νευρώνων (FGFR1/ FGF8), τη μετανάστευσή τους από την εμβρυϊκή τους καταβολή προς τον υποθάλαμο (KAL1, PROK2/ PROKR2), τη ρύθμιση της έκκρισης της GnRH, (KISS1/ KISS1R, ΤAC3/TACR3) και τη σύνδεσή της με τον υποδοχέα της (GNRHR) (Σχήμα 2). Οι μεταλλάξεις στον GNRHR προκαλούν κλινική εικόνα που κυμαίνεται από μερική έως πλήρη αντίσταση στην GnRH και ευθύνεται για περίπου το 50% των οικογενών περιπτώσεων ΙΥΥ και ένα μικρό ποσοστό σποραδικών περιπτώσεων 8,21. Τέλος, πρόσφατα βρέθηκε ότι περιπτώσεις μεμονωμένου ΙΥΥ οφείλονται σε μεταλλάξεις της GnRH 2,22,23.

Σχήμα 2: Σχηματική αναπαράσταση των γονιδίων που συμμετέχουν στην οντογένεση και λειτουργία του υποθαλαμο–υποφυσιακο-γοναδικού άξονα (Bonomi Μ, et al. Asian Journal of Andrology. 2012;14(1):49–56)
Η παραδοσιακή θεώρηση ότι το σύνδρομο Kallmann και ο ΙΥΥ οφείλονται σε μονογονιδιακή διαταραχή έχει πλέον αναθεωρηθεί. Η σύγχρονη αντίληψη είναι ότι αποτελούν ένα σύνθετο γενετικό νόσημα με ποικιλία στην έκφραση, τη διεισδυτικότητα και τον τύπο κληρονομικότητας. Η παρουσία μεταλλάξεων σε δύο γονίδια (δι- γονιδιακές) ή σε περισσότερα (ολιγο-γονιδιακές) στο ίδιο άτομο με ΙΥΥ μπορεί πλέον να εξηγήσει την ποικιλομορφία στο φαινότυπο που απαντάται στα μέλη της ίδιας οικογένειας.
Επιπλέον, στην παθογένειά του ενδέχεται να συμμετέχουν και περιβαλλοντικοί παράγοντες, οι οποίοι μπορεί να ασκούν επιγενετική δράση στην έκφραση των γονιδίων με αποτέλεσμα την εμφάνιση πολυμορφισμών ή άλλων γενετικών ελλειμμάτων σε πολλαπλά γονίδια 15,17,18,24.
Κλινική εικόνα
Το σύνδρομο Kallmann απαντάται στο 60% ενώ, ο νορμο-οσμικός ΥΥ στο 40% των περιπτώσεων ασθενών με συγγενή ανεπάρκεια της GnRH21. Η συγγενής ανεπάρκεια της GnRH αφορά σε συντριπτική πλειοψηφία τους άνδρες (πενταπλάσιο ποσοστό σε σχέση με τις γυναίκες) και μπορεί να εκδηλωθεί σε οποιαδήποτε ηλικία. Η κλινική εικόνα είναι ανάλογη με την ηλικία εκδήλωσης της νόσου 9,25.
- Νεογνική περίοδος: Τα αγόρια εμφανίζονται με μικρούς όρχεις, μικροφαλλία και / ή κρυψορχία (λόγω ανεπαρκούς δράσης του άξονα κατά την εμβρυϊκή ζωή).
- Παιδική ηλικία: Κατά τη διάρκεια αυτής της περιόδου ο υποθαλαμο–υποφυσιακο-γοναδικός άξονα παραμένει αδρανής, οπότε δεν υπάρχουν σημεία υπογοναδισμού. H διάγνωση κλινικά μπορεί να γίνει μόνο στην περίπτωση ύπαρξης ανοσμίας (Kallmann) ή παρουσίας συνοδών συγγενών ανωμαλιών πχ. λαγώχειλο, λυκόστομα, συνδακτυλία, συν-κινησία άκρων και ετερόπλευρη αγενεσία νεφρού 21.
- Εφηβεία: Χαρακτηρίζεται από καθυστέρηση ήβης, με φυσιολογικό ανάστημα στην πλειονότητα των περιπτώσεων αλλά, ευνουχοειδείς αναλογίες (ανάπτυγμα χεριών ≥ ανάστημα + 2-5εκ.) λόγω καθυστέρησης στη σύγκλειση των επιφύσεων ενώ ο όγκος των όρχεων παραμένει στα προεφηβικά επίπεδα (<4ml).
- Ενήλικες: Ειδική μνεία θα πρέπει να γίνει στον ΙΥΥ των ενηλίκων (adult onset IHH). Πρόκειται για σπάνια επίκτητη μορφή ανεπάρκειας της GnRH, που εκδηλώνεται για πρώτη φορά στην ενήλικο ζωή ως υπογονιμότητα και σεξουαλική δυσλειτουργία. Εμφανίζεται σε άνδρες με φυσιολογική ενήβωση και προηγούμενη σεξουαλική και αναπαραγωγική λειτουργία (συμπεριλαμβανομένης και της τεκνοποίησης) 25,26,27,28.
Διάγνωση
Η διάγνωση βασίζεται στην κλινική υποψία της ύπαρξης υπογοναδισμού και στην περίπτωση του σ. Kallmann στην επιπρόσθετη παρουσία ανοσμίας και άλλων συνοδών ανωμαλιών χαρακτηριστικών του συνδρόμου. Στην περίπτωση που η κλινική υποψία τίθεται λόγω της καθυστέρησης της εμφάνισης της ήβης είναι απαραίτητη η διαφορική διάγνωση από την ιδιοσυστασιακή καθυστέρηση της ήβης η οποία είναι πολύ συχνότερη (βλ παρακάτω). Στην περίπτωση αυτή η διάγνωση του ΥΥ μπορεί με ασφάλεια να τεθεί μόνο σε ηλικία μεγαλύτερη των 18 ετών 29. Τα χαμηλά επίπεδα τεστοστερόνης ορού (<100 mg/ dl) σε συνδυασμό με χαμηλά ή απρόσφορα φυσιολογικά επίπεδα γοναδοτροπινών θέτουν τη διάγνωση του δευτεροπαθούς υπογοναδισμού. Κατά τη διάρκεια όμως της παιδικής ηλικίας, οπότε οι τιμές γοναδοτροπινών και στεροειδών του φύλου είναι φυσιολογικά χαμηλές ή μη ανιχνεύσιμες, η ανίχνευση επίσης χαμηλών συγκεντρώσεων αντι-μυλλέριας ορμόνης (ΑΜΗ) και ανασταλτίνης Β (οι οποίες υπό φυσιολογικές συνθήκες είναι αυξημένες) είναι διαγνωστικές του ΥΥ στην παιδική ηλικία (σχήμα 1) καθιστώντας περιττή τη χρήση δοκιμασιών διέγερσης (GnRH test) για την αιτιολογική προσέγγιση του υπογοναδισμού στα αγόρια 29 .
Κατά τη διερεύνηση της αιτιολογίας του δευτεροπαθούς υπογοναδισμού απαραίτητη είναι η απεικόνιση της περιοχής υποθαλάμου-υπόφυσης (μαγνητική τομογραφία) για τον αποκλεισμό χωροκατακτητικής εξεργασίας ή άλλης διηθητικής νόσου. Στο σύνδρομο Kallmann, η MRI εγκεφάλου μπορεί να δείξει ετερόπλευρη ή αμφοτερόπλευρη απουσία ή υποπλασία των οσφρητικών βολβών. Ο απεικονιστικός έλεγχος θα πρέπει να συμπληρώνεται με υπερηχογράφημα όρχεων και νεφρών (για πιθανή αγενεσία νεφρών) ενώ αναγκαίος είναι και ο ορμονολογικός έλεχγος της επάρκειας των υπόλοιπων υποφυσιακών ορμονών. Με τον αποκλεισμό των άλλων αιτίων του δευτεροπαθούς υπογοναδισμού, τίθεται η διάγνωση του ιδιοπαθούς υπογοναδοτροπικού υπογοναδισμού ή του συνδρόμου Kallmann (όταν συνυπάρχει ανοσμία).
Τέλος, το λεπτομερές ιστορικό (κληρονομικό αναμνηστικό) για την ύπαρξη στην οικογένεια ατόμων με ανοσμία, απουσία ή καθυστέρηση της ήβης, υπογοναδισμό ή υπογονιμότητα είναι απαραίτητο καθώς τόσο το σύνδρομο Kallmann και όσο και ο ΙΥΥ έχουν γενετική βάση 8.
Θεραπεία
Η θεραπευτική παρέμβαση εξαρτάται από την ηλικία και το θεραπευτικό στόχο που μπορεί να είναι, η επαγωγή των δευτερογενών χαρακτηριστικών του φύλου και η αύξηση του μεγέθους των έξω γεννητικών οργάνων, η αντιμετώπιση του υπογοναδισμού ή η επαγωγή της σπερματογένεσης για την αντιμετώπιση της υπογονιμότητας.
Η χορήγηση τεστοστερόνης αποτελεί την κύρια θεραπευτική παρέμβαση καθώς επιτυγχάνει τόσο την επαγωγή των δευτερογενών χαρακτηριστικών του φύλου και την αύξηση του μεγέθους του πέους όσο και την αντιμετώπιση των συμπτωμάτων και των συνεπειών του υπογοναδισμού. Καθώς όμως, η τεστοστερόνη δεν επιδρά στον όγκο των όρχεων συχνά προτιμάται να προηγείται η χορήγηση χοριακής γοναδοτροπίνης (hCG) στην εφηβεία, μόνη ή σε συνδυασμό με FSH, καθώς η τελευταία είναι απαραίτητη για την ωρίμανση των κυττάρων Sertoli και του σπερματικού επιθηλίου, σκοπεύοντας στην όσο το δυνατόν καλύτερη ανάπτυξή τους και την εξασφάλιση καλύτερου δυναμικού για μελλοντική γονιμότητα19. Για την αντιμετώπιση της υπογονιμότητας μπορεί να χρησιμοποιηθεί η χορήγηση GnRH μέσω αντλίας υποδόριας έγχυσης (εάν πρόκειται για υποθαλαμικής αιτιολογίας υπογοναδισμό) ή (το συνηθέστερο) θεραπεία με γοναδοτροπίνες: ανθρώπινη χοριακή γοναδοτροπίνη (hCG) γοναδοτροπίνες από εμμηνοπαυσμένες γυναίκες (hMG) ή rFSH 19,30 .
Ανεξάρτητα από τη θεραπευτική παρέμβαση ο παραγόμενος αριθμός σπερματοζωαρίων είναι συνήθως πολύ χαμηλότερος του φυσιολογικού. Εν τούτοις, επιτυγχάνεται κύηση στο 50-80% των περιπτώσεων με αριθμό σπερματοζωαρίων 5εκ/ ml 30,31. Σύμφωνα με μελέτες οι παράγοντες που επηρεάζουν την επιτυχή έκβαση της θεραπείας είναι: ο αρχικός όγκος όρχεων, η απουσία ιστορικού κρυψορχίας, η προηγηθείσα λήψη γοναδοτροπινών, η μη προηγηθείσα λήψη τεστοστερόνης και τιμές ανασταλτίνης Β >60pg/ml 32.
Η αποκατάσταση επαρκούς σπερματογένεσης μπορεί να είναι μια χρονοβόρα διαδικασία και ο χρόνος αναμονής για την επίτευξη κύησης ποικίλλει από 6-9 μήνες έως 2 χρόνια 30,31,33. Οι τεχνικές υποβοηθούμενης αναπαραγωγής (ART) έχουν θέση στις περιπτώσεις που δεν επιτευχθεί κύηση μετά από 20 μήνες θεραπείας ή 8 μήνες μετά από την επίτευξη συγκέντρωσης σπέρματος 5εκ/ml ή εφόσον υπάρχει γυναικείος παράγοντας υπογονιμότητας 34.
Φυσική ιστορία – Πρόγνωση
Παλαιότερα ο ΙΥΥ και το σύνδρομο Kallmann θεωρούνταν μη αναστρέψιμη διαταραχή, η οποία απαιτούσε θεραπεία υποκατάστασης με στεροειδή εφ΄όρου ζωής. Φαίνεται, όμως, από δεδομένα της τελευταίας εικοσαετίας 35,36,37,38 ότι υπάρχουν περιπτώσεις αυτόματης αποκατάστασης της λειτουργίας του υποθαλαμο-υποφυσιακο-γοναδικού άξονα (Reversal IHH) και κατά συνέπεια της στεροειδο-γένεσης και/ ή της σπερματογένεσης σε ποσοστό 5 – 22% 39,36,37, ενώ ένα ποσοστό αυτών εμφανίζει υποτροπή του υπογοναδοτροπικού υπογοναδισμού (relapse) μετά από έκθεση σε στρες (συναισθηματικό, ψυχιατρικό, μεταβολικό) 36.
Συνεπώς, συστήνεται πλέον η δια βίου παρακολούθηση των ασθενών και η εκτίμηση του όγκου των όρχεων για την ανίχνευση περιπτώσεων αυτόματης αποκατάστασης του άξονα. Προτείνεται ακόμα και η δοκιμαστική διακοπή της θεραπείας υποκατάστασης για διάστημα 3-6 μηνών μετά την ολοκλήρωση της ήβης και ακολούθως ο προσδιορισμός της τεστοστερόνης και των γοναδοτροπινών 30.
3. Σύνδρομο Prader – (Labhart) – Willi
Πρόκειται για σπάνια γενετική διαταραχή με συχνότητα εμφάνισης 1: 10.000 – 1: 30.000. Οφείλεται σε μικρές απαλείψεις στο πατρικό χρωμόσωμα 15, στην περιοχή 15q11-q13, τουλάχιστον στο 70% των περιπτώσεων ενώ, το 25% οφείλεται σε μητρική δισωμία του χρωμοσώματος 15 και το 5% σε άλλες διαταραχές 39.
Κλινική εικόνα: Το σύνδρομο έχει χαρακτηριστικό φαινότυπο, που περιλαμβάνει: σοβαρή υποτονία του νεογνού, υπερφαγία, νοσογόνο παχυσαρκία, κοντό ανάστημα με κοντά άνω και κάτω άκρα, προβλήματα συμπεριφοράς, μαθησιακές δυσκολίες και υπογοναδισμό 39.
Ο υπογοναδισμός είναι μικτής αιτιολογίας. Οφείλεται δηλαδή, σε υποθαλαμική διαταραχή με ανεπαρκή έκκριση της GnRH αλλά και σε πρωτοπαθή ανεπάρκεια των όρχεων. Συνήθως εκδηλώνεται με μικροφαλλία, υποπλαστικό όσχεο και κρυψορχία ενώ μέχρι σήμερα δεν έχει αναφερθεί στη βιβλιογραφία καμία περίπτωση πατρότητας40. Στην πλειονότητα των ασθενών εμφανίζεται καθυστερημένη ή ατελής ήβη, αν και σε ένα ποσοστό περίπου 4% μπορεί να εμφανιστεί πρώιμη ήβη και στα δύο φύλα. Στο 15-30% μπορεί να εκδηλωθεί πρώιμη αδρεναρχή, λόγω της παχυσαρκίας ή λόγω της αυξημένης έκθεσης των επινεφριδίων σε ινσουλίνη και IGF-1 8,40,41.
Διάγνωση: Επί κλινικής υποψίας σε άτομο με υπογοναδισμό, ήπια νοητική υστέρηση, υπερφαγία, και κεντρική παχυσαρκία θα πρέπει να επιβεβαιώνεται η διάγνωση με μοριακή- γενετική ανάλυση 8.
Θεραπεία: Στους περισσότερους ασθενείς απαιτείται χορήγηση ορμονικής θεραπείας για την πρόκληση και τη διατήρηση της ήβης. Ο υπογοναδισμός στους άνδρες αντιμετωπίζεται με τη χορήγηση τεστοστερόνης, ωστόσο, χρειάζεται στενή παρακολούθηση λόγω της πιθανής δυσμενούς επίδρασης του στεροειδούς στη συμπεριφορά του ασθενούς 41,42,43.
4. Σύνδρομα Bardet-Biedl και Laurence-Moon
Είναι σπάνιες αυτοσωματικά κληρονομούμενες υπολειπόμενες γενετικές διαταραχές με παρόμοιο φαινότυπο που περιλαμβάνει μελαγχρωστική αμφιβληστροειδοπάθεια, κεντρική παχυσαρκία, πνευματική καθυστέρηση, δομικές και λειτουργικές διαταραχές των νεφρών και υπογοναδισμό. Διαφοροποιούνται από την παρουσία πολυδακτυλίας στο σύνδρομο Bardet-Biedl και νευρολογικών διαταραχών όπως αταξία και σπαστική παραπληγία και απουσία πολυδακτυλίας στο σύνδρομο Laurence-Moon. Δεν έχει ακόμη διευκρινιστεί αν πρόκειται για διαφορετικές εκφράσεις της ίδιας διαταραχής ή για διαφορετικές κλινικές οντότητες 44 (εικόνα 1) .

Εικόνα 1. Σύνδρομο Bardet-Biedl (Singh KK. et al, 2015, Indian J Nephrol.2015; 25(5):300-302).
Το σύνδρομο Bardet-Biedl είναι συχνότερο από το σύνδρομο Laurence-Moon με επίπτωση 1 στις 125.000–160.000 γεννήσεις στην Ευρώπη και 1 /65.000 στον αραβικό πληθυσμό 44. Παρουσιάζει μεγάλη ετερογένεια. Μέχρι τώρα είναι γνωστές 19 μεταλλάξεις γονιδίων (BBS γονίδια) που θεωρούνται υπεύθυνες για το 80% των περιπτώσεων. Τα γονίδια αυτά βρίσκονται στα χρωμοσώματα 2, 3, 4, 7, 8, 9, 11, 12, 14, 15, 16 και 17 8,45. Η γενετική βάση του συνδρόμου Laurence-Moon δεν είναι γνωστή.
Ο υπογοναδισμός είναι βασική διαταραχή των συνδρόμων αυτών. Αν και παλιότερα θεωρούνταν υπογοναδοτροπικός, σήμερα πιστεύεται ότι υπάρχει πρωτοπαθής βλάβη των όρχεων. Συνήθως, παρατηρείται μικροφαλλία και μικροί όρχεις, κρυψορχία (9%), καθυστέρηση της ενήβωσης ενώ έχει αναφερθεί και ατροφία σπερματικών σωληναρίων και ποικίλου βαθμού αναστολή της σπερματογένεσης σε μερικές περιπτώσεις. Εντούτοις, έχουν αναφερθεί σποραδικές περιπτώσεις πατρότητας σε άνδρες με σύνδρομο Bardet-Biedl 8,46.
5. Παρεγκεφαλιδική αταξία και υπογοναδοτροπικός υπογοναδισμός
Είναι κλινικά και γενετικά ετερογενής ομάδα διαταραχών, κληρονομούμενες με υπολειπόμενο αυτοσωματικό χαρακτήρα, των οποίων η γενετική βάση δεν έχει πλήρως διαλευκανθεί47. Το σύνδρομο Gordon Holmes είναι η πρώτη από τις διαταραχές αυτές η οποία έχει περιγραφεί από τον Holmes το 190848 και χαρακτηρίζεται από παρεγκεφαλιδική αταξία τύπου Holmes, υπογοναδοτροπικό υπογοναδισμό, νυσταγμό και πιθανή πνευματική καθυστέρηση. Το σύνδρομο Boucher-Neuhäuser μπορεί να θεωρηθεί συνέχεια του συνδρόμου Gordon Holmes και χαρακτηρίζεται από παρεγκεφαλιδική αταξία, υπογοναδοτροπικό υπογοναδισμό και χορειοαμφιβληστροειδική δυστροφία. Τα νευρολογικά χαρακτηριστικά εμφανίζονται προοδευτικά τη 2η και 3η δεκαετία της ζωής. Ο υπογοναδοτροπικός υπογοναδισμός εκδηλώνεται συνήθως με απουσία ενήβωσης και οφείλεται σε βλάβη των υποφυσιακών κυττάρων τα οποία αδυνατούν να ανταποκριθούν στη δράση της GnRH ή εμφανίζουν μερική απάντηση ενώ υπάρχουν και περιστατικά με υποθαλαμική βλάβη και φυσιολογική απάντηση στην GnRH 48,49.
6. Συγγενής υποπλασία επινεφριδίων και υπογοναδοτροπι-κός υπογοναδισμός
Είναι σπάνια φυλοσύνδετη νόσος η οποία κληρονομείται με τον υπολειπόμενο χαρακτήρα. Οφείλεται σε μετάλλαξη του γονιδίου NR0B1 που εδράζεται στο βραχύ σκέλος του Χ χρωμοσώματος στην περιοχή Xp21.3–21.2. και κωδικοποιεί την πρωτεΐνη DAX-1 (Dosage-sensitive sex reversal – Adrenal hypoplasia congenital critical region on the X chromosome 1) η οποία είναι ένας ορφανός πυρηνικός υποδοχέας 8.
Η κλασσική μορφή της νόσου χαρακτηρίζεται από πρωτοπαθή επινεφριδιακή ανεπάρκεια, υπογοναδοτροπικό υπογοναδισμό και υπογονιμότητα. Στο 40 % των περιπτώσεων η επινεφριδιακή ανεπάρκεια εκδηλώνεται τους δύο πρώτους μήνες της ζωής με απώλεια άλατος λόγω της έλλειψης τόσο των αλατοκορτικοειδών όσο και των γλυκοκορτικοειδών. Στις υπόλοιπες περιπτώσεις εμφανίζεται ηπιότερα κατά τη διάρκεια της παιδικής ηλικίας. Ο υπογοναδοτροπικός υπογοναδισμός εκδηλώνεται με απουσία εμφάνισης ήβης ή σπανιότερα με διακοπή της εξέλιξής της και φαίνεται να οφείλεται τόσο σε υποθαλαμική όσο και υποφυσιακή βλάβη η οποία οδηγεί σε διαταραχή της έκκρισης των γοναδοτροπινών η οποία δεν εμφανίζεται στα πρώτα χρόνια της ζωής. Εκτός από την κλασσική μορφή της νόσου υπάρχουν περιπτώσεις στις οποίες προεξάρχει η ανεπάρκεια των αλατοκορτικοειδών και άλλοτε η ανεπάρκεια γλυκοκορτικοειδών γεγονός που δημιουργεί διαφοροδιαγνωστικά προβλήματα. Άλλα χαρακτηριστικά της νόσου μπορεί να είναι η μυϊκή δυστροφία του Duchenne ή ανεπάρκεια της γλυκερολικής κινάσης 50.
Περιστασιακά η νόσος μπορεί να πρωτοεμφανιστεί σε νεαρούς ενήλικες (late onset) και χαρακτηρίζεται από ιστορικό προοδευτικής ή ήπιας επινεφριδιακής ανεπάρκειας και μερικό υπογοναδοτροπικό υπογοναδισμό.
Η αντιμετώπιση της κλασικής μορφής της νόσου περιλαμβάνει τη θεραπεία υποκατάστασης με αλατοκορτικοειδή και γλυκοκορτικοειδή και επαγωγή της ήβης είτε με χορήγηση hCG είτε τεστοστερόνης 50.
7. Μεμονωμένη ανεπάρκεια LH ή FSH
Η μεμονωμένη ανεπάρκεια της LH εμφανίστηκε για πρώτη φορά στη διεθνή βιβλιογραφία το 1950 με τον όρο «γόνιμος ευνούχος» λόγω της χαρακτηριστικής εικόνας της συνύπαρξης του υπογοναδισμού με φυσιολογικό μέγεθος όρχεων και παρουσία φυσιολογικών σπερματοζωαρίων σε ικανοποιητική ποσότητα 51. Ο εργαστηριακός έλεγχος είναι χαρακτηριστικός με χαμηλά επίπεδα LΗ και τεστοστερόνης και φυσιολογικά επίπεδα FSH ενώ η βιοψία όρχεως δείχνει ολιγοζωοσπερμία με φυσιολογική ποιότητα σπέρματος και ατροφία κυττάρων Leydig. Η αιτία της μεμονωμένης ανεπάρκειας της LH δεν έχει πλήρως διευκρινισθεί ενώ σε μία περίπτωση διαπιστώθηκε μετάλλαξη του γονιδίου του υποδοχέα της GnRHR. Στην περίπτωση αυτή η μακροχρόνια χορήγηση hCG είχε σαν αποτέλεσμα την ομαλοποίηση των επιπέδων τεστοστερόνης και της σπερματογένεσης ενώ μετά τη διακοπή της διαπιστώθηκε αποκατάσταση του υπογοναδισμού με διατήρηση φυσιολογικών επιπέδων τεστοστερόνης 52.
Μόνο τρεις περιπτώσεις ανδρών έχουν αναφερθεί με αδρανοποιητικές μεταλλάξεις τoυ γονιδίου της β-υπομονάδας της LH, ομόζυγες ή διπλά ετερόζυγες, οι οποίοι εμφάνιζαν υπογοναδισμό, καθυστέρηση της ήβης και μικρό μέγεθος όρχεων, χαμηλά ή μη ανιχνεύσιμα επίπεδα της LH και αυξημένα επίπεδα FSH λόγω της δευτεροπαθούς διαταραχής στην ανάπτυξη των κυττάρων Sertoli, ενώ στη βιοψία όρχεων διαπιστώθηκε διακοπή της σπερματογένεσης στο στάδιο της μείωσης και απουσία κυττάρων Leydig. Οι πάσχοντες είναι πλήρως αρρενοποιημένοι κατά τη γέννηση, λόγω της δράσης της hCG κατά τη διάρκεια της ενδομήτριας ζωής, και ο υπογοναδισμός εμφανίζεται ως απουσία ενήβωσης. 8, 28,53,54.
Μεταλλάξεις του γονιδίου της β-υπομονάδας της FSH οδηγούν σε μεμονωμένη ανεπάρκειά της. Μέχρι στιγμής πολύ λίγα περιστατικά έχουν αναφερθεί στη διεθνή βιβλιογραφία και χαρακτηρίζονται από ελαττωμένο αριθμό κυττάρων Sertoli, αζωοσπερμία με φυσιολογική ενήβωση και επίπεδα τεστοστερόνης φυσιολογικά ή στα κατώτερα φυσιολογικά όρια ενώ σε μία περίπτωση παρατηρήθηκε απουσία εμφάνισης ήβης και χαμηλά επίπεδα τεστοστερόνης 55.
Έχουν περιγραφεί και περιπτώσεις με ανεπάρκεια FSH χωρίς μεταλλάξεις του γονιδίου της β-υπομονάδας της η οποία εκδηλώθηκε με διαταραχή της σπερματογένεσης και πλήρη αρρενοποίηση. Η αιτιολογία της ανεπάρκειας παραμένει άγνωστη 56.
8. Συγγενής υποϋποφυσισμός με παρουσία υπογοναδοτροπικού υπογοναδισμού
Είναι σπάνιες διαταραχές που χαρακτηρίζονται από μεταλλάξεις γονιδίων μεταγραφικών παραγόντων οι οποίοι είναι υπεύθυνοι για την οντογένεση και ανάπτυξη της υπόφυσης. Η πιο συχνή από αυτές είναι εκείνη του μεταγραφικού παράγοντα PROP 1 η οποία οδηγεί σε ανεπάρκεια αυξητικής ορμόνης, προλακτίνης, θυρεοειδοτρόπου ορμόνης και γοναδοτροπινών ενώ μπορεί να συνυπάρχει και ανεπάρκεια κορτικοτρόπου ορμόνης. Πρόκειται για αυτοσωματική υπολειπόμενη διαταραχή. Ο χρόνος έναρξης και η βαρύτητα της ανεπάρκειας των υποφυσιακών ορμονών ποικίλει. Οι περισσότεροι ασθενείς παρουσιάζουν πρώιμη εμφάνιση της ανεπάρκειας της GH και της TSH με αποτέλεσμα κοντό ανάστημα και υποθυρεοειδισμό ενώ η κλινική εικόνα της έλλειψης των γοναδοτροπινών κυμαίνεται από απουσία εμφάνισης ήβης έως αυτόματη ενήβωση και εμφάνιση του υπογοναδισμού αργότερα στην ενήλικο ζωή. Οι ίδιες ανεπάρκειες υποφυσιακών ορμονών παρατηρούνται και στις περιπτώσεις αδρανοποιητικών μεταλλάξεων του γονιδίου LHX3 χωρίς όμως ανεπάρκεια της ACTH, οι οποίες συνοδεύονται επιπλέον από κοντή και άκαμπτη αυχενική μοίρα της σπονδυλικής στήλης με περιορισμένη δυνατότητα περιστροφής της κεφαλής και κινήσεων του κορμού 57.
Η διαφραγματο-οπτική δυσπλασία είναι μία σπάνια ετερογενής διαταραχή η αιτιοπαθογένεια της οποίας αποδίδεται σε μεταλλάξεις του γονιδίου HEX 1 ή άλλων γνωστών (SOX3) ή άγνωστων γονιδίων και περιβαλλοντικούς παράγοντες καθώς η συχνότητα ανεύρεσης μεταλλάξεων του HEX1 στη διαταραχή αυτή είναι πολύ μικρή. Χαρακτηρίζεται από την παρουσία τουλάχιστον δύο από τις τρεις παρακάτω διαταραχές: διαταραχές μέσης γραμμής, απλασία οπτικού νεύρου και υποϋποφυσισμό. Η κλινική εικόνα ποικίλει ενώ 62% των ασθενών παρουσιάζουν κάποιου βαθμού υποφυσιακή ανεπάρκεια, χωρίς να παρατηρείται σε όλες τις περιπτώσεις υπογοναδοτροπικός υπογοναδισμός 58.
Επίσης, έχει περιγραφεί μία μορφή φυλοσύνδετου υποϋποφυσισμού που συνοδεύεται από άλλοτε άλλου βαθμού πνευματική καθυστέρηση και αποδίδεται σε διπλασιασμό της περιοχής Xq26.1-q27.3 στην οποία βρίσκεται το γονίδιο SOX3. Τέλος, υποπλασία του πρόσθιου λοβού της υπόφυσης με υπογοναδοτροπικό υπογοναδισμό σε συνδυασμό με αμφοτερόπλευρη ανοφθαλμία/μικροφθαλμία, οισοφαγική ατρησία και νευροαισθητηριακή απώλεια ακοής έχει παρατηρηθεί σε de novo μεταλλάξεις του γονιδίου SOX2 57,58.
9. Επίκτητος υπογοναδοτροπικός υπογoναδισμός
Οποιαδήποτε ανατομική ή λειτουργική βλάβη στην περιοχή του υποθαλαμο-υποφυσιακού άξονα μπορεί να προκαλέσει υπογοναδοτροπικό υπογοναδισμό είτε λόγω διαταραχής της παραγωγής ή/και της έκκρισης της GnRH είτε λόγω διαταραχής της παραγωγής ή/και της έκκρισης των γοναδοτροπινών ή λόγω συνδυασμού των ανωτέρω. Η βαρύτητα του υπογοναδισμού εξαρτάται από το σημείο και την έκταση της βλάβης και συνήθως συνυπάρχει με ανεπάρκεια και άλλων υποφυσιακών ορμονών ενώ σπανιότερα μπορεί να εκδηλωθεί ως μεμονωμένος υπογοναδοτροπικός υπογοναδισμός 8.
Χωροκατακτητικές εξεργασίες στην περιοχή του υποθαλάμου και της υπόφυσης είναι από τις συχνότερες αιτίες (πίνακας 1). Τα αδενώματα της υπόφυσης μέσω της πίεσης προκαλούν βλάβη στα γοναδοτρόπα κύτταρα της υπόφυσης ενώ τα προλακτινώματα, επιπρόσθετα αναστέλλουν την παραγωγή και την έκκριση των γοναδοτροπινών μέσω της υπερπρολακτιναιμίας. Τα κρανιοφαρυγγιώματα, μηνιγγιώματα ή άλλοι πρωτοπαθείς ή μεταστατικοί όγκοι του κεντρικού νευρικού συστήματος μπορεί να προκαλέσουν διαταραχή στην παραγωγή της GnRH και των γοναδοτροπινών είτε λόγω πίεσης του μίσχου είτε λόγω της διήθησης των κυττάρων του υποθαλάμου και της υπόφυσης. Οι κοκκιωματώδεις νόσοι όπως σαρκοείδωση, ιστιοκύττωση και φυματίωση καθώς και η λεμφοκυτταρική υποφυσίτιδα μπορούν επίσης να προσβάλλουν τον υποθάλαμο και την υπόφυση και να προκαλέσουν υπογοναδοτροπικό υπογοναδισμό. Με τον ίδιο μηχανισμό δρα και η αιμοχρωμάτωση η οποία μπορεί να οδηγήσει σε προοδευτική ανεπάρκεια των γοναδοτροπινών. Συστηματικές παθήσεις όπως δρεπανοκυτταρική και μεσογειακή αναιμία, κίρρωση ήπατος, χρόνια νεφρική ανεπάρκεια μπορεί να προκαλέσουν υπογοναδοτροπικό υπογοναδισμό ενώ παθήσεις του γαστρεντερικού όπως η νόσος Crohn έχουν ενοχοποιηθεί για καθυστέρηση της εμφάνισης της ήβης. Διαταραχές της θρέψης όπως συμβαίνει στα σύνδρομα δυσαπορρόφησης, νευρογενή ανορεξία, καχεξία προκαλούν διαταραχή στην υποθαλαμική έκκριση της GnRH στην οποία οφείλεται και ο υπογοναδισμός που εμφανίζεται σε αθλητές λόγω υπέρμετρης άσκησης. Επίσης, λοιμώξεις του ΚΝΣ, κατάγματα της βάσης του κρανίου και αιμορραγικές ή αποφρακτικές βλάβες στην περιοχή του υποθαλάμου ή ακτινοθεραπεία όγκων του εγκεφάλου μπορεί να προκαλέσουν διαταραχή στην υποθαλαμο-υποφυσιακή λειτουργία. Τέλος, πολύ συχνή αιτία υπογοναδοτροπικού υπογοναδισμού αποτελούν φάρμακα όπως αναβολικά στεροειδή, κορτικοστεροειδή, οπιούχα και ψυχοτρόπα φάρμακα όπως οι φαινοθειαζίδες τα οποία καταστέλλουν τον υποθαλαμο-υποφυσιακό άξονα 8,59.
Εκτός από τις παραπάνω περιπτώσεις επίκτητου υπογοναδοτροπικού υπογοναδισμού έχει αναγνωριστεί μία σπάνια διαταραχή στην έκκριση των γοναδοτροπινών σε ενήλικες στους οποίους έχει αποκλειστεί οποιαδήποτε ανατομική ή συστηματική ή λειτουργική διαταραχή ενώ δεν έχει βρεθεί καμία γενετική διαταραχή που να προκαλεί υπογοναδοτροπικό υπογοναδισμό στους ασθενείς αυτούς 28.
Η κλινική εικόνα εξαρτάται από την ηλικία εμφάνισης, τη βαρύτητα του υπογοναδισμού και την υποκείμενη νόσο.
Η διάγνωση θα βασιστεί κυρίως στο ιστορικό, στην κλινική εικόνα και στον εργαστηριακό έλεγχο με μέτρηση των γοναδοτροπινών και της πρωινής τεστοστερόνης, τον προσδιορισμό των επιπέδων των άλλων υποφυσιακών ορμονών για τον αποκλεισμό της πολλαπλής υποφυσιακής ανεπάρκειας, την εκτίμηση της θυρεοειδικής λειτουργίας και την ανίχνευση της παρουσίας υπερπρολακτιναιμίας, τη μέτρηση των επιπέδων φερριτίνης και στον απεικονιστικό έλεγχο κυρίως με μαγνητική τομογραφία της περιοχής του υποθαλάμου και της υπόφυσης. Ο απεικονιστικός έλεγχος θα πρέπει να γίνεται στις περιπτώσεις σοβαρού υπογοναδισμού (επίπεδα τεστοστερόνης < 150 ng/dl), πολλαπλής υποφυσιακής ανεπάρκειας, επιμένουσας υπερπρολακτιναιμίας και τοπικών συμπτωμάτων ενδεικτικών παρουσίας χωροκατακτητικής εξεργασίας (κεφαλαλγία, διαταραχές οπτικών πεδίων και όρασης κλπ )1,11,59.
Η θεραπεία βασίζεται στην αντιμετώπιση της υποκείμενης νόσου, όταν αυτό είναι εφικτό, και στη χορήγηση θεραπείας ορμονικής υποκατάστασης με τεστοστερόνη ή επαγωγή της σπερματογένεσης με χορήγηση γοναδοτροπινών ή GnRH (σπανιότερα) όταν είναι επιθυμητή η γονιμότητα 1.
10. Υπερπρολακτιναιμία
Η προλακτίνη είναι μία πεπτιδική ορμόνη που εκκρίνεται από τα λακτοτρόπα κύτταρα του πρόσθιου λοβού της υπόφυσης. Παρόλο που έχουν βρεθεί υποδοχείς προλακτίνης στα κύτταρα Leydig, στα σπερματοκύτταρα, στις σπερματίδες, στον προστάτη, στην επιδιδυμίδα, στους σπερματοδόχους πόρους και στις σπερματοδόχους κύστεις, εντούτοις η φυσιολογική της δράση στο γεννητικό σύστημα στον ενήλικα άνδρα είναι άγνωστη, ενώ έχει επιβεβαιωθεί μόνο η ανασταλτική δράση της εκκρινόμενης προλακτίνης μετά τον οργασμό στη σεξουαλική επιθυμία και συμπεριφορά 60,61. Η έκκριση της προλακτίνης ελέγχεται από τον υποθάλαμο κυρίως ανασταλτικά με την έκκριση της ντοπαμίνης αλλά και διεγερτικά με την έκκριση των εκλυτικών παραγόντων της προλακτίνης (PRF) TSH, βαζοπρεσίνη και VIP (σχήμα 3).

Σχήμα 3. Παράγοντες που ρυθμίζουν την έκκριση της προλακτίνης ( Vyas U. BJMP 2012;5(4):a534).
Αιτιοπαθογένεια: Οποιαδήποτε διαταραχή στην παραγωγή, μεταφορά και δράση της ντοπαμίνης στα λακτοτρόπα κύτταρα της υπόφυσης ή η διέγερση των λακτοτρόπων κυττάρων από τους διεγερτικούς παράγοντες μπορεί να οδηγήσει σε υπερπρολακτιναιμία (πίνακας 3).
Ήπια αύξηση των επιπέδων προλακτίνης μπορεί να προκληθεί από σωματικό ή ψυχολογικό στρες. Τα προλακτινώματα αποτελούν τη συχνότερη αιτία υπερπρολακτιναιμίας και ανάλογα με το μέγεθός τους διακρίνονται σε μικροαδενώματα (διάμετρος<10 χιλιοστά) και μακροαδενώματα (διάμετρος >10 χιλιοστά). Συνήθως είναι σποραδικά, αν και έχουν αναφερθεί και οικογενείς περιπτώσεις, ενώ τα κακοήθη προλακτινώματα είναι σπάνια. Τα περισσότερα αποτελούνται εξ ολοκλήρου από λακτοτρόπα κύτταρα με εξαίρεση ένα μικρό ποσοστό που είναι μικτά και αποτελούνται συνήθως από λακτοτρόπα και σωματρόπα κύτταρα. Τα επίπεδα της προλακτίνης είναι συνήθως ανάλογα με το μέγεθος του αδενώματος.
Άλλοι όγκοι της υπόφυσης που δεν εκκρίνουν προλακτίνη, κατά κύριο λόγο μακροαδενώματα, κρανιοφαρυγγιώματα και όγκοι του μίσχου της υπόφυσης, προκαλούν υπερπρολακτιναιμία λόγω πίεσης του μίσχου η οποία οδηγεί σε διαταραχή της πυλαίας κυκλοφορίας και κατά συνέπεια της μεταφοράς της ντοπαμίνης στην υπόφυση. Επίσης, διαταραχές του υποθαλάμου όπως όγκοι, αρτηριοφλεβώδεις δυσπλασίες, φλεγμονώδη και διηθητικά νοσήματα (σαρκοείδωση, ιστιοκύττωση) μέσω της ελαττωμένης έκκρισης και απελευθέρωσης της ντοπαμίνης μπορεί να οδηγήσουν σε υπερπρολακτιναιμία. Επιπλέον, βλάβες του υποθαλάμου μπορεί να προκαλέσουν έκκριση των εκλυτικών παραγόντων της προλακτίνης και κατά συνέπεια υπερπρολακτιναιμία. Ο υποθυρεοειδισμός είναι επίσης σημαντική αιτία υπερπρολακτιναιμίας όπως και η χρόνια νεφρική ανεπάρκεια και η κίρρωση του ήπατος 8, 62.
Φαρμακευτικοί παράγοντες που ανταγωνίζονται τη δράση της ντοπαμίνης στους D2 υποδοχείς της στα λακτοτρόπα κύτταρα της υπόφυσης όπως οι φαινοθειαζίνες, οι βουτυροφαινόνες, η ιμιπραμίνη και οι βενζαμίδες (μετοκλοπραμίδη, δομπεριδόνη, σουλπιρίδιο), αναστέλλουν τη σύνθεση της ντοπαμίνης όπως η μεθυλντόπα, αναστέλλουν την απελευθέρωση της ντοπαμίνης όπως η ρεζερπίνη ή διεγείρουν απευθείας την παραγωγή και έκκριση της προλακτίνης όπως οι Η2 αναστολείς και τα οιστρογόνα είναι συχνά αίτια υπερπρολακτιναιμίας (πίνακας 4) 8, 62.
Η υπερπρολακτιναιμία προκαλεί ελάττωση της κατά ώσεις έκκρισης της GnRH με αποτέλεσμα τη διαταραχή της έκκρισης των γοναδοτροπινών η οποία οδηγεί σε υπογοναδισμό που εκφράζεται με μειωμένα επίπεδα τεστοστερόνης και καταστολή της σπερματογένεσης. Επίσης, προκαλεί μείωση των εκκριτικών αιχμών της LH η οποία οδηγεί σε ελάττωση της έκκρισης της τεστοστερόνης. Επιπλέον, αναστέλλει τη μετατροπή της τεστοστερόνης σε διυδροτεστοστερόνη 62,63.
Πίνακας 3. Αίτια υπερπρολακτιναιμίας
| Φυσιολογικά |
| Άσκηση |
| Ύπνος |
| Στρες |
| Παθολογικά |
| Βλάβη υποθαλάμου- μίσχου υπόφυσης |
| Κοκκιωματώδεις νόσοι |
| Διηθητικά νοσήματα |
| Ακτινοβολία |
| Κύστη του Rathke |
| Τραύμα |
| Όγκοι: κρανιοφαρυγγίωμα, γερμίνωμα, μεταστάσεις στον υποθάλαμο, μηνιγγίωμα, επέκταση υποφυσιακής μάζας στον υπερεφιππιακό χώρο |
| Βλάβη στην υπόφυση |
| Προλακτίνωμα |
| Μεγαλακρία |
| Λεμφοκυτταρική υποφυσίτιδα-επέκταση μάζας στον παραεφιππιακό χώρο |
| Μακροαδένωμα |
| Μακροπρολακτιναιμία |
| Επέμβαση |
| Τραύμα |
| Ιδιοπαθής |
| Συστηματικές διαταραχές |
| Χρόνια νεφρική ανεπάρκεια |
| Κίρρωση ήπατος |
| Επιληπτικές κρίσεις |
| Ακτινοβολία κρανίου |
| Νευρογενής |
| Ερεθισμός θωρακικού τοιχώματος με τραύμα, επέμβαση, έρπης ζωστήρας |
| Φαρμακευτικά αίτια |
Κλινική εικόνα: Η υπερπρολακτιναιμία στους άνδρες εκδηλώνεται κυρίως με υπογοναδοτροπικό υπογοναδισμό. Η σεξουαλική δυσλειτουργία είναι η συχνότερα εμφανιζόμενη διαταραχή ( 88%) και περιλαμβάνει τη στυτική δυσλειτουργία η οποία τις περισσότερες φορές συνυπάρχει με μειωμένη libido. Σε μερικές περιπτώσεις έχει αναφερθεί παλίνδρομη εκσπερμάτιση. Ελάττωση της τριχοφυΐας (40%), γυναικομαστία (21%) και γαλακτόρροια (13%) αποτελούν σπανιότερες εκδηλώσεις της υπερπρολακτιναιμίας. Με τα ως άνω συμπτώματα μπορεί να συνυπάρχουν και άλλα, ενδεικτικά της αιτίας της υπερπρολακτιναιμίας, όπως είναι συμπτώματα προκαλούμενα από πίεση του μίσχου (κεφαλαλγία, διαταραχές της όρασης και των οπτικών πεδίων). Όμως, μπορεί ορισμένοι ασθενείς με υπερπρολακτιναιμία να παραμένουν ασυμπτωματικοί 8, 62.
Διάγνωση: Βασίζεται στην ανεύρεση αυξημένων επιπέδων προλακτίνης. Ο προσδιορισμός των επιπέδων προλακτίνης ακολουθείται από εκείνον των άλλων υποφυσιακών ορμονών όταν πιθανολογείται διαταραχή της υποφυσιακής λειτουργίας.
Προσοχή χρειάζεται στην ερμηνεία των αυξημένων επιπέδων τις προλακτίνης. Η πιθανότητα της μακροπρολακτιναιμίας καθώς επίσης και της υπερπρολακτιναιμίας της οφειλόμενης σε στρες θα πρέπει να αποκλειστούν στις περιπτώσεις που χαρακτηρίζονται από απουσία συμπτωματολογίας ενδεικτικής υπερπρολακτι-ναιμίας. Το στρες προκαλεί ήπια αύξηση των επιπέδων προλακτίνης που δεν ξεπερνούν το διπλάσιο του ανώτερου φυσιολογικού ορίου.
Οι ασθενείς με επαναλαμβανόμενα αυξημένα επίπεδα προλακτίνης, προσδιοριζόμενα υπό κατάλληλες συνθήκες και εφόσον έχει αποκλεισθεί η πιθανότητα της φαρμακευτικής υπερπρολακτιναιμίας, θα πρέπει να διερευνώνται περαιτέρω με απεικονιστικό έλεγχο της περιοχής του υποθαλάμου και της υπόφυσης με μαγνητική τομογραφία προκειμένου να αποκλεισθεί η πιθανότητα προλακτινώματος ή άλλης χωροκατακτητικής εξεργασίας του υποθαλάμου ή της υπόφυσης.
Θεραπεία: Εξαρτάται από την αιτία της υπερπρολακτιναιμίας. Οι αγωνιστές της ντοπαμίνης (βρωμοκρυπτίνη, κιναγολίδη, καμπεργολίδη) αποτελούν τον ακρογωνιαίο λίθο της αντιμετώπισης των προλακτινωμάτων. Επιτυγχάνουν μείωση των επιπέδων της προλακτίνης και του μεγέθους του αδενώματος, αύξηση των επιπέδων τεστοστερόνης και αντιμετώπιση των συμπτωμάτων της υπερπρολακτιναιμίας (στυτική δυσλειτουργία, μειωμένη libido, υπογονιμότητα). Ο πλέον χρησιμοποιούμενος φαρμακευτικός παράγοντας είναι η καμπεργολίδη λόγω του μεγάλου χρόνου ημίσειας ζωής που την καθιστά αποτελεσματική χορηγούμενη 1-2 φορές εβδομαδιαίως. Η χειρουργική θεραπεία εφαρμόζεται στο 10% περίπου των προλακτινωμάτων τα οποία δεν απαντούν στη φαρμακευτική αγωγή και στις περιπτώσεις υπερπρολακτιναιμίας οφειλόμενης σε άλλους όγκους της υπόφυσης και του υποθαλάμου.
Στις περιπτώσεις που παρατηρείται υπογοναδισμός παρά την επιτυχή αντιμετώπιση της υπερπρολακτιναιμίας με τους αγωνιστές της ντοπαμίνης είναι απαραίτητη η θεραπεία υποκατάστασης με τεστοστερόνη. Επίσης, στις περιπτώσεις στις οποίες προκαλείται υποφυσιακή ανεπάρκεια λόγω χειρουργικής αντιμετώπισης ή ακτινοθεραπείας εκτός από υποκατάσταση με τεστοστερόνη απαιτείται και υποκατάσταση με θυροξίνη και κορτιζόνη.
Η υπερπρολακτιναιμία που οφείλεται σε στρες ή στην παρουσία μακρο-προλακτίνης δεν χρειάζεται αντιμετώπιση.
Πίνακας 4. Φάρμακα που προκαλούν υπερπρολακτιναιμία
| Οπιοειδή, Μεθαδόνη |
| Ψυχοτρόπα φάρμακα |
| Νευροληπτικά |
| Βενζαμίδες: αμισουλπιρίδη, σουλπιρίδη, σουλτοπρίδη, τριαπρίδη |
| Φαινοθειαζίνες: χλωροπρομαζίνη, φλουφεναζίδη, λεβομεπρομαζίδη, φαιρφεναζίδη, πιπετιαζίνη, προπερικιαζίνη, θειριδαζίνη |
| Βουτυροφαινόνες: Δροπεριδόλη, αλοπεριδόλη, πιπαμπερόνη, φλουπεντιξόλη, λοξαπίνη, ολανζαπίνη, πιμποζίνη, ρισπεριδόνη, ζουκλοπεντιξόλη |
| Τρικυκλικά αντικαταθλιπτικά: αμιτρυπτιλίνη, αμοξαπίνη, χλωριμιπραμίνη, δεσιπραμίνη, δοσουλιπίνη, δοξεπίνη, ιμιπραμίνη, μαπροτιλίνη, τριμιπραμίνη |
| Αντιεμετικά: μετοκλοπραμίδη, μετοπιμαζίδη |
| Η2 αναστολείς: σιμετιδίνη |
| Αντιυπερτασικά: ρεζερπίνη, α-μεθυλ-ντόπα |
| Οιστρογόνα |
11. Βιβλιογραφία
1. Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, and Montori VM. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010; 95(6):2536–2559.
2. Balasubramanian R and Crowley WF Jr. Isolated GnRH deficiency : a disease model serving as a unique prism into the systems biology of the GnRH neuronal network. Mol Cell Endocrinol. 2011; 346:4-12
3. Nachtigall LB, Boepple PA, Pralong FP and Crowley WF Jr. Adult-onset idiopathic hypogonadotropic hypogonadism—a treatable form of male infertility. N Engl J Med. 1997; 336: 410–15.
4. Rosner W, Auchus RJ, Azziz R, Sluss PM and Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab. 2007; 92: 405–13.
5. Bhasin S and Basaria S. Diagnosis and treatment of hypogonadism in men. Best Pract Res Clin Endocrinol Metab. 2011;25:251–270
6. Grinspon, R.P., Loreti, N., Braslavsky, D., Valeri, C., Schteingart, H., Ballerini, M.G. Bedecarrás P, Ambao V, Gottlieb S, Ropelato MG, Bergadá I, Campo SM and Rey RA. Spreading the clinical window for diagnosing fetal-onset hypogonadism in boys. Front Endocrinol. 2014;5:51.
7. Basaria S. Male hypogonadism. Lancet. 2014; 383: 1250–63.
8. Hermann M. Behre, Eberhard Nieschlag, Carl-Joachim Partsch, Peter Wieacker, and Manuela Simoni. Diseases of the Hypothalamus and the Pituitary Gland . Andrology Nieschlag E, Behre H, Nieschlag S Eds, 3rd edition, Springer. 2010; pp. 193-238.
9. Fraietta R, Zylberstjn DS and Esteves SC . Hypogonadotropic Hypogonadism Revisited. Clinics (Sao Paulo). 2013 Feb; 68(S1): 81-88.
10. Popa SM, Clifton DK andSteiner RA. The role of kisspeptins and GPR54 in the neuroendocrine regulation of reproduction. Annu Rev Physiol. 2008; 70: 213-38.
11. Young J. Approach to the Male Patient with Congenital Hypogonadotropic Hypogonadism . J Clin Endocrinol Metab. 2012; 97(3):707–718.
12. Mitchell AL, Dwyer A, Pitteloud N and Quinton R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab. 2011;22:249–258.
13. Schwanzel-Fukuda M and Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989; 338: 161–4.
14. Wray S, Grant P andGainer H. Evidence that cells expressing luteinizing hormone releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci USA. 1989; 86: 8132–6.
15. Bonomi Μ, Libri DV, Guizzardi F, Guarducci E, Maiolo E, Pignatti E, Asci R and Persani L. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian Journal of Andrology. 2012;14(1):49–56.
16. Balasubramanian R, Dwyer A, Seminara SB, Pitteloud N, Kaiser UB and Crowley WFJr. Human GnRH deficiency : a unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinology. 2010; 92: 81-99.
17. Sykiotis GP, Plummer L, Hughes VA , Durrani S, Nayak-Young S, Dwyer AA, Quinton R, Hall JE, Gusella JF, Seminara SB, Crowley WF Jr and Pitteloud N. Oligogenic basis of isolated gondotropin- releasing hormone deficiency. Proc Natl Acad Sci USA. 2010; 107: 15140- 15144.
18. Valdes-Socin H, Almanza MR, Fernández-Ladreda MT, Debray FG, Bours V and Beckers A. Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front. Endocrinol. July 2014;5 article 109.
19. Boehm U., Bouloux PM, Dattani M, de Roux N, Dode C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R and Young J. Expert consensus document: European consensus statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015;11:547-564.
20. Lewkowitz – Shpuntoff HM, Hughes VA, Plummer L, Au MG, Doty RL, Seminara SB, Chan YM, Pitteloud N, Crowley WFJr and Balasubramanian R. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism : pathophysiological and genetic implications. J Clin Endocrinol Metab. 2012;97:Ε 136- Ε 144.
21. Shin SJ, Sul Y, Kim JH, Cho JH, Kim GH, Kim JH, Choi JH and Yoo HW. Clinical, endocrinological, and molecular characterization of Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism : a single center experience. Ann Pediatr Endocrinol Metab. 2015;20:27-33.
22. Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombès M, Millar RP, Guiochon-Mantel A and Young J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–2748
23. Chan YM, de Guillebon A, Lang-Muritano M, Plummer L, Cerrato F, Tsiaras S, Gaspert A, Lavoie HB, Wu CH, Crowley WF Jr, Amory JK, Pitteloud N and Seminara SB. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 2009;106:11703–11708.
24. Brioude F, Bouligand J,Trabado S,Francou B,Salenave S,Kamenicky P, Brailly-Tabard S, Chanson P, Guiochon-Mantel A and Young J. Non-syndromic congenital hypogonadotropic hypogonadism: clinical presentation and genotype-phenotype relationships. Eur J Endocrinol. 2010;162:835–851.
25. UpToDate 2015.
26. Cerrato F, Shagoury J, Kralickova M, Dwyer A, Falardeau J, Ozata M, Van Vliet G, Bouloux P, Hall JE, Hayes FJ, Pitteloud N, Martin KA, Welt C and Seminara SB.et al. Coding sequence analysis of GNRHR and GPR54 in patients with congenital and adult-onset forms of hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2006; 155Suppl 1: S3–10.
27. Nachtigall LB, Boepple PA, Pralong FP and Crowley WF Jr. Adult-onset idiopathic hypogonadotropic hypogonadism—a treatable form of male infertility. N Engl J Med. 1997; 336: 410–15.
28. Dwyer AA, Hayes FJ, Plummer L, Pitteloud N and Crowley WF Jr. The long-term clinical follow-up and natural history of men with adult-onset idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2010;95:4235–4243.
29. Rey RA, Grinspon RP, Gottlieb S, Pasqualini T, Knoblovits P, Aszpis S, Pacenza N, Usher S, Bergada I and Campo SM. Male hypogonadism: an extended classification based on developmental endocrine physiology-based approach. Andrology. 2013;1:3-16.
30. Han TS and Bouloux PM. What is the optimal therapy for young males with hypogonadotrpic hypogonadism? Clin Endocrinol (Oxf). 2010; 72(6):731-737.
31. ZitzmannM and Nieschlag E. Hormone substitution in male hypogonadism. Mol Cell Endocrinol. 2000; 161 (1-2): 73-88.
32. King T and Hayes FJ. Long- term outcome of idiopathic hypogonadotropic hypogonadism. Curr Opin Endocrinol Diabetes Obes. 2012;19:204–210.
33. Resorlu B, Abdulmajed MI, Kara C, Unsal A and Aydos K. Is intracytoplasmic sperm injection essential for the treatment of hypogonadotropic hypogonadism? Α comparison between idiopathic and secondary hypogonadotrophic hypogonadism. Hum Fertil (Camb). 2009;12(4):204-8.
34. Bacircioglu ME, Erden HF, Ciray HN, Bayazit N and Bahceci M. Gonadotrophin therapy in combination with ICSI in men with hypogonadotrophic hypogonadism. Reprod Biomed Online. 2007;15(2):156-60.
35. Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF Jr and Pitteloud N. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 2007; 357: 863–73.
36. Sidhoum VF, Chan YM, Lippincott MF, Balasubramanian R., Quinton R, Plummer L, Dwyer A, Pitteloud N, Hayes FJ, Hall JE, Martin KA, Boepple BA and Seminara SB. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab 2014; 99: 861-870.
37. Mao JF, Xu HL, Duan J, Chen RR, Li L, Li B, Nie M, Min L, Zhang HB and Wu XY. Reversal of idiopathic hypogonadotropic hypogonadism: a cohort study in Chinese patients. Asian Journal of Andrology (2015) 17, 497–502.
38. Pitteloud N, Acierno JS Jr, Meysing AU, Dwyer AA, Hayes FJ, and Crowley Jr WF Jr. Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab. 2005; 90: 1317–22.
39. KY Jin. Systematic review of the clinical and genetic aspects of Prader-Willi syndrome. Korean J Pediatr. 2011;54(2):55-63
40. Vogels A, Moerman P, Frijns JP and Bogaert GA. Testicular histology in boys with Prader-Willi syndrome: fertile or infertile? J Urol. 2008;180(4):1800–1804.
41. Emerick and Vogt. Endocrine manifestations and management of Prader – Willi syndrome. International Journal of Pediatric Endocrinology. 2013; 2013:14
42. Gross-Tsur V, Hirsch HJ, Benarroch F and Eldar-Geva T. The FSH – Inhibin axis in Prader-Willi syndrome: heterogeneity of gonadal dysfunction. Reproductive Biology and Endocrinology. 2012;10:39
43. Eldar-Geva T, Hirsch HJ, Benarroch F, Rubinstein O and Gross-Tsur V. Hypogonadism in females with Prader- Willi syndrome from infancy to adulthood: variable combinations of a primary gonadal defect and hypothalamic dysfunction. European Journal of Endocrinology. 2010;162: 377-384.
44. Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, Stefanelli M, Murphy C, Cramer BC, Dean JCS, Beales PL, Katsanis N, Bassett AS, Davidson WS, and Parfrey PS. Clinical and Genetic Epidemiology of Bardet–Biedl Syndrome in Newfoundland: A 22-Year Prospective, Population-Based, Cohort Stud. Am J Med Genet A. 2005. Feb 1; 132(4): 352–360.
45. Forsythe E and Beales PL. Bardet–Biedl syndrome. European Journal of Human Genetics. 2013; 21: 8–13
46. Forsythe E and Beales PL. Bardet-Biedl Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2003 Jul 14 (updated 2015 Apr 23).
47. Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A, Hannequin D, Strom TM, Prokisch H, Kernstock C, Durr A, Schöls L, Lima-Martínez MM, Farooq A, Schüle R, Stevanin G, Marques W Jr and Züchner S. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014 Jan;137 (Pt 1):69-77.
48. Topaloglu AK, Lomniczi A, Kretzschmar D, Dissen GA, Kotan LD, McArdle CA, Koc AF, Hamel BC, Guclu M, Papatya ED, Eren E, Mengen E, Gurbuz F, Cook M, Castellano JM, Kekil MB, Mungan NO, Yuksel B and Ojeda SR. Loss-of-function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome. J Clin Endocrinol Metab. 2014 Oct;99(10):E2067-75.
49. Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, Hall JE, Adam I, Dwyer A, Plummer L, Aldrin SV, O’Rourke J, Kirby A, Lage K, Milunsky A, Milunsky JM, Chan J, Hedley-Whyte ET, Daly MJ, Katsanis N and Seminara SB. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013 May 23;368(21):1992-2003.
50. Suntharalingham JP, Buonocore F, Duncan AJ and Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Practice & Research Clinical Endocrinology & Metabolism 29 (2015) 607e619
51. Pasqualini RQ and Burr GE. Sindrome hypoandrogenico con gametogenesis conservada. Classifi cation del la insuffi ciencia testicular. Rev Asoc Med Argent 1950;64:6–19
52. Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF Jr and Hayes FJ. The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab. 2001;86:2470–2475
53. Shiraishi K and Naito K. Fertile Eunuch Syndrome with the Mutations (Trp8Arg and Ile15Thr) in the b subunit of Luteinizing Hormone. Endocrine Journal 2003;50: 733–737
54. Huhtaniemi I and Alevizaki M. Mutations along the hypothalamic–pituitary–gonadal axis affecting male reproduction. Reproductive BioMedicine Online. 2007; 15(6): 622-632.
55. Lofrano-Porto A, Casulari LA, Nascimento PP, Giacomini L, Naves LA, da Motta LD and Layman LC. Effects of follicle-stimulating hormone and human chorionic gonadotropin on gonadal steroidogenesis in two siblings with a follicle-stimulating hormone beta subunit mutation. Fertil Steril. 2008;90:1169– 1174.
56. Mantovani G, Borgato S, Beck-Peccoz P, Romoli R, Borretta G and Persani L. Isolated follicle-stimulating hormone (FSH) deficiency in a young man with normal virilization who did not have mutations in the FSH beta gene. Fertil Steril. 2013;79:434–436
57. Kelberman and Dattani MT. The role of transcription factors implicated in anterior pituitary development in the aetiology of congenital hypopituitarism. Annals of Medicine. 2006; 38: 560–577.
58. Mehta A and Dattani MT. Developmental disorders of the hypothalamus and pituitary gland associated with congenital hypopituitarism. Best Practice & Research Clinical Endocrinology & Metabolism. 2008; 22(1):191–206.
59. Silveira LF and Latronico AC. Approach to Hypogonadotropic Hypogonadism. Clin Endocrinol Metab, May 2013, 98(5):1781–1788
60. Andrzej Bartke. Prolactin in the Male: 25 Years Later. Journal of Andrology. 2004; 25( 5): 661–836
61. Krüger THC, Paake P, Haverkamp J, Kramer M, Exton MS, Saller B, Leygraf N, Hartmann U and Schedlowski M. Effects of acute prolactin manipulation on sexual drive and function in males. J Endocrinol. 2003;179: 357–365.
62. Buvat J. Hyperprolactinemia and sexual function in men: a short review. International Journal of Impotence Research. 2003;15, 373–377.
63. Lobo RA and Kletzky OA. Normalization of androgen and sex-hormone-binding globulin levels after treatment of hyperprolactinemia. J Clin Endocr Metab 1982; 56: 562–566.
Created: 11 November 2019
Last update: 11 November 2019