
Στέλιος Φουντουλάκης
Ειδικευόμενος Ενδοκρινολόγος, Διδάκτωρ Πανεπιστημίου Ιωαννίνων,
Ενδοκρινολογική Κλινική και Διαβητολογικό Κέντρο Γ.Ν.Α «Γ. Γεννηματάς»,
Αγαθοκλής Τσατσούλης
Καθηγητής Ενδοκρινολογίας-Παθολογίας, Διευθυντής Ενδοκρινολογικής
Κλινικής –Διαβητολογικού Κέντρου Πανεπιστημιακού Νοσοκομείου Ιωαννίνων
Εισαγωγή
Η θυρεοειδική αυτοανοσία (ΘΑ) αποτελεί τη συχνότερη οργανοειδική αυτοάνοση διαταραχή, προσβάλλοντας έως και 3% του γενικού πληθυσμού (επίπτωση: 4,3/1000/έτος στις γυναίκες, 0,88/1000/έτος στους άνδρες) (1). Οι διαφορετικές εκφάνσεις της ΘΑ περιλαμβάνουν τη νόσο του Graves (GD) και στον αντίποδα τη θυρεοειδίτιδα Hashimoto (ΗΤ) και τις υποκατηγορίες της. Παρόλο που το τελικό αποτέλεσμα της GD και της HT, στη θυρεοειδική λειτουργία, είναι εκ διαμέτρου αντίθετο, οι δύο αυτές νόσοι εμφανίζουν πολλά κοινά χαρακτηριστικά με αποτέλεσμα την υπόθεση περί συγγενούς αιτιολογικής βάσης. Τέτοια χαρακτηριστικά είναι: 1) H παρουσία αυτοαντισωμάτων έναντι κοινών θυρεοειδικών αντιγόνων (θυρεοειδικής υπεροξειδάσης/anti-TPO και θυρεοσφαιρίνης / anti-Tg). 2) Η διήθηση του θυρεοειδούς από Τ λεμφοκύτταρα τα οποία στρέφονται εναντίον θυρεοειδικών αντιγόνων. 3) Η συνύπαρξη με άλλα αυτοάνοσα νοσήματα. 4) Η δυνατότητα μετάπτωσης της μιας νόσου στην άλλη κατά τη φυσική τους εξέλιξη.
Η θυρεοειδική αυτοανοσία είναι μια σύνθετη ομάδα διαταραχών στην αιτιοπαθογένεια των οποίων εμπλέκονται γενετικοί (80%) και περιβαλλοντικοί παράγοντες (20%). Αποτέλεσμα της επίδρασης των παραπάνω παραγόντων αποτελεί η ενεργοποίηση ανοσολογικών μηχανισμών που αρχικά οδηγούν στη διαφυγή ορισμένων κυττάρων της λευκής σειράς (υποκατηγορίες Τ και Β λεμφοκυττάρων) από τους μηχανισμούς ανοσολογικής ανοχής και έπειτα στην είσοδο στο θυρεοειδή όπου και αλληλεπιδρούν με τα θυρεοκύτταρα. Στην περίπτωση της ΗΤ και των παραλλαγών της, η διατάραξη της ισορροπίας μεταξύ θυρεοκυττάρων και ενδοθυρεοειδικών λεμφοκυττάρων, οδηγεί σε διήθηση του θυρεοειδούς από Τh1/Th17 λεμφοκύτταρα, εμφάνιση anti-TPO / anti-Tg αντισωμάτων, καταστροφή του θυρεοκυττάρου και υποθυρεοειδισμό, ενώ στην GD σε διήθηση του θυρεοειδούς από Τh2 (και λιγότερο Th1) λεμφοκύτταρα, παραγωγή anti-TPO / anti-Tg αντισωμάτων και διεγερτικών αντισωμάτων έναντι του υποδοχέα της TSH (thyroid stimulating antibodies/ΤSAb) με επακόλουθο υπερθυρεοειδισμό (2). Άλλα αυτοαντισώματα που ανιχνεύονται σε ασθενείς με ΘΑ είναι αυτά έναντι του συμμεταφορέα νατρίου-ιωδίου (NIS) o οποίος εδράζεται στη βασικοπλάγια (basolateral) πλευρά των θυρεοειδικών κυττάρων του θυλακίου και έναντι της pendrin η οποία συντελεί στην ενδοκυττάρια μεταφορά του ιωδίου προς την κορυφαία (apical) πλευρά του θυρεοκυττάρου (10% στην GD και 8% στην HT σε σειρά ασθενών στη Μ. Βρετανία) (3).
Οι μελέτες που ερευνούν τις αυτοάνοσες θυρεοειδικές νόσους εστιάζουν στον καθορισμό της αιτιοπαθογένειάς τους, με σκοπό την ανάπτυξη αιτιολογικών θεραπευτικών μέσων στη θέση των συμπτωματικών που εφαρμόζονται σήμερα. Στο κεφάλαιο αυτό γίνεται αναφορά στις νεότερες εξελίξεις που αφορούν στους μηχανισμούς οι οποίοι εμπλέκονται στην αιτιοπαθογένεια της ΘΑ.
1. Κατηγοριοποίηση θυρεοειδικής αυτοανοσίας
Η ταξινόμηση της ΘΑ περιλαμβάνει τους διάφορους τύπους της χρόνιας αυτοάνοσης θυρεοειδίτιδας (θυρεοειδίτιδα Hashimoto-HT) και τη GD.
- Η ΗΤ χαρακτηρίζεται από την παρουσία βρογχοκήλης, θυρεοειδικών αυτοαντισωμάτων (κυρίως anti-TPO και anti-Tg) και δυσλειτουργίας του θυρεοειδούς. Θεωρείται αποτέλεσμα καταστροφής των θυρεοειδικών κυττάρων από άφθονα αυτοαντιδραστικά Τ λεμφοκύτταρα (T effector cells) που διηθούν το θυρεοειδή και τα οποία μέχρι πρόσφατα θεωρούνταν του Th1 τύπου, ως επί το πλείστον (4). Νεότερες μελέτες έχουν δείξει πως εμπλέκονται τόσο Τh2 όσο και Τh17 λεμφοκύτταρα, ο ρόλος των οποίων προσφάτως αποτέλεσε αντικείμενο έρευνας (5). Το τελικό αποτέλεσμα είναι ο υποθυρεοειδισμός.
- Στα αρχικά στάδια η ΗΤ μπορεί να εμφανίζεται ως υποκλινικός υποθυρεοειδισμός, ο οποίος φαίνεται πως αποτελεί το αρχικό στάδιο της θυρεοειδίτιδας Hashimoto αλλά παρατηρείται και στη σιωπηλή θυρεοειδίτιδα. Είναι πιο συχνά αυτοάνοσης αιτιολογίας και χαρακτηρίζεται από θετικό τίτλο αντιθυρεοειδικών αντισωμάτων, φυσιολογικές τιμές θυρεοειδικών ορμονών και οριακά υψηλά επίπεδα TSH (>5 μU/ml). Σε νεαρά άτομα, επίπεδα της TSH μεγαλύτερα από 3,5 μU/ml, θεωρούνται παθολογικά. Ασθενείς με αυξημένο τίτλο αντισωμάτων και υποκλινικό υποθυρεοειδισμό εξελίσσονται σε υποθυρεοειδική κλινική ΗΤ με συχνότητα 5% ανά έτος. Ο επιπολασμός του υποκλινικού υποθυρεοειδισμού είναι 7,5-8,5% στις γυναίκες και 3-4,4% στους άνδρες ενώ αυξάνει με την ηλικία, προσεγγίζοντας το 17 % σε ηλικίες άνω των 60 ετών. Στη μελέτη NHANES III η συχνότητα εμφάνισής του ήταν 5,8% σε γυναίκες και 3,4% σε άνδρες άνω των 12 ετών ενώ στη Whickman ήταν 4-5% σε γυναίκες 18-44 ετών και 17,4% σε άνω των 70 (6,7).
- Υποκατηγορία της ΗΤ θεωρείται και η σπάνια θυρεοειδίτιδα Riedel (Riedel struma), η οποία χαρακτηρίζεται από αντικατάσταση του θυρεοειδικού ιστού από ινώδη. Η ίνωση μπορεί να εκτείνεται και πέραν του θυρεοειδούς με συμμετοχή των παρακείμενων μυών και ιστών. Θεωρείται πως ίσως αποτελεί τελικό στάδιο της ΗΤ (8).
- Η ατροφική θυρεοειδίτιδα συνοδεύεται από κλινικό υποθυρεοειδισμό και χαρακτηρίζεται από έναν μικρό και ατροφικό θυρεοειδή με λεμφοκυτταρική διήθηση και παρέγχυμα που έχει αντικατασταθεί από ινώδη ιστό. Ανταγωνιστικά αντισώματα εναντίον του υποδοχέα της TSH βρίσκονται στο 20-50% των ασθενών με ατροφική θυρεοειδίτιδα σε σχέση με 10% σε ασθενείς με ΗΤ, υποδηλώνοντας πιθανή εμπλοκή στην αιτιοπαθογένεια της νόσου (9).
- Άλλη μορφή της αυτοάνοσης θυρεοειδίτιδας αποτελεί η σιωπηλή (ανώδυνη) θυρεοειδίτιδα με τις υποδιαιρέσεις της: τη σποραδική και τη θυρεοειδίτιδα της λοχείας. Και οι δύο είναι παροδικές αλλά μπορεί να επανεμφανιστούν.
- Πρόσφατα μια νέα κατηγορία ΗΤ με ίνωση, η ΙgG4-θυρεοειδίτιδα, έχει περιγραφεί από τους ερευνητές. Οι περισσότεροι τη θεωρούν ως εντελώς ξεχωριστή κλινική οντότητα, που ίσως ομοιάζει με τα αρχικά στάδια της ΗΤ, ενώ κάποιοι θεωρούν πως αλληλεπικαλύπτεται με τη θυρεοειδίτιδα Riedel (10). Χαρακτηρίζεται από υπεργαμμασφαιριναιμία, αυξημένο αριθμό ΙgG4 κυττάρων στο περιφερικό αίμα, ενδοθυρεοειδική ίνωση, διάχυτη υποηχογένεια, μικρότερη ηλικία εμφάνισης, υψηλότερο επίπεδο αντιθυρεοειδικών αντισωμάτων, μικρότερη αναλογία γυναικών/ανδρών και πιο συχνή εμφάνιση υποκλινικού υποθυρεοειδισμού σε σχέση με την ΗΤ (11). Μπορεί να εμφανιστεί στα πλαίσια μιας νέας κλινικής οντότητας που ονομάζεται Νόσος σχετιζόμενη με IgG4 (ΙgG4-related disease). Η νόσος αυτή μπορεί να περιλαμβάνει ογκόμορφες βλάβες στο πάγκρεας, τους δακρυϊκούς αδένες και τις χοληφόρες οδούς, οπισθοπεριτοναϊκή ίνωση, σκληρυντική χολαγγειίτιδα και σιαλαδενίτιδα, ψευδοόγκους ήπατος και οφθαλμικού κόγχου, λεμφοειδική διάμεση πνευμονία, αυτοάνοση παγκρεατίτιδα, σωληναριοδιάμεση νεφρίτιδα, παχυμηνιγγίτιδα και υποφυσίτιδα. Οι κατευθυντήριες οδηγίες για τη διάγνωση του νέου αυτού συνδρόμου δημοσιεύθηκαν πρόσφατα στη συνάντηση του Research Program for Intractable Disease στην Ιαπωνία (12).
Τέλος η GD οφείλεται.σε διεγερτικά αντισώματα του υποδοχέα της TSH (TSAb) και χαρακτηρίζεται από υπερπλασία των θυρεοειδικών θυλακίων, μικρότερη λεμφοκυτταρική διήθηση απ’ότι η ΗΤ (κυρίως Τh2 λεμφοκύτταρα) και παρουσία αυτοαντισωμάτων (2). Στα αρχικά στάδια μπορεί να εμφανίζεται ως υποκλινικός υπερθυρεοειδισμός με φυσιολογικές τιμές θυρεοειδικών ορμονών και TSH χαμηλότερη του φυσιολογικού. Η συχνότητα του υποκλινικού υπερθυρεοειδισμού κυμαίνεται από 0,7-12,4% και ο ρυθμός εξέλιξης του σε κλινικό υπερθυρεοειδισμό είναι 5% ανά έτος (13,14).
2. Ανοσολογική απάντηση και αυτοανοσία
Προτού αναλυθεί ο σημαντικός ρόλος του ανοσολογικού συστήματος στην αιτιοπαθογένεια της ΘΑ, κρίνεται σκόπιμη η εν συντομία παρουσίαση των βασικών αρχών λειτουργίας του όσον αφορά στην αντιγονοπαρουσίαση, τις κατηγορίες λεμφοκυττάρων (CD4, CD8, B, Tregs, Th17, Th22, Th23, Tfh) την παραγωγή κυτοκινών και την ενεργοποίηση/αλληλεπίδρασή τους με τα αντιγονοπαρουσιαστικά κύτταρα (antigen presenting cells/APC) καθώς και των μηχανισμών ανοσολογικής ανοχής και διαφυγής από αυτήν.
2.1. Κατηγορίες κυττάρων του ανοσοποιητικού συστήματος με ρόλο στην ανάπτυξη ΘΑ
Η βασική λειτουργία του ανοσοποιητικού συστήματος είναι η διάκριση των αυτοαντιγόνων από τα εξωγενή αντιγόνα και η εξουδετέρωση των τελευταίων. Για την επίτευξη του στόχου αυτού σημαντικός είναι ο ρόλος των CD4 T βοηθητικών λεμφοκυττάρων, τα οποία μετά από αντιγονική επαφή, διαφοροποιούνται σε δύο υποκατηγορίες, τα Th1 και τα Th2 λεμφοκύτταρα. Τα Th1 εκκρίνουν κυρίως ιντερλευκίνη-2 (IL-2), ιντερφερόνη-γ (IFN-γ) και παράγοντα νέκρωσης των όγκων (TNF-α), ρυθμίζοντας την κυτταρική ανοσία, ενώ τα Th2 εκκρίνουν κυρίως ιντερλευκίνη 4,5,6 και 10, επηρεάζοντας τη χυμική ανοσία (παραγωγή αντισωμάτων από B λεμφοκύτταρα) (15). Αντίθετα τα CD8 κυτταροτοξικά Τ λεμφοκύτταρα συνήθως εκφράζουν αντιγονοειδικό υποδοχέα TCR ο οποίος ενεργοποιείται από αντιγόνα τα οποία παρουσιάζονται από μόρια του μείζονος συμπλέγματος ιστοσυμβατότητας MHC-I που εκφράζονται στην επιφάνεια των APC. Τα CD8 Τ λεμφοκύτταρα εμπλέκονται στην καταστροφή των κυττάρων που φέρουν τα συγκεκριμένα αντιγόνα, μέσω κυτοτοξινών όπως περφορίνες και granzyme. Για την αντιγονοπαρουσίαση απαραίτητη είναι η πρόσδεση του αντιγόνου σε μόρια MHC-I και ΙΙ της επιφάνειας των ΑPC και στη συνέχεια η παρουσίασή του στους TCR υποδοχείς της επιφάνειας των CD8 και CD4 λεμφοκυττάρων αντίστοιχα. Τα APC (περιλαμβάνονται δενδριτικά, μακροφάγα και Β κύτταρα) ενδοκυτταρώνουν αντιγόνα και στη συνέχεια τα παρουσιάζουν μέσω των μορίων MHC-Ι και ΙΙ που φέρουν στην επιφάνειά τους. Ιδιαίτερη σημασία έχει το γεγονός πως ορισμένα ενεργοποιημένα επιθηλιακά κύτταρα, μεταξύ των οποίων και τα θυρεοκύτταρα, μπορούν να δρουν ως APC παρουσιάζοντας αντιγόνα στα Τ λεμφοκύτταρα. Επίσης η ΙL23 που παράγεται από δενδριτικά και μακροφάγα μπορεί να ενεργοποιεί την αυτοάνοση απάντηση μέσω μιας νεοπεριγραφείσας κατηγορίας λεμφοκυττάρων, των Th17 (2,16).
Η ενεργοποίηση των Τ λεμφοκυττάρων απαιτεί, εκτός του σήματος που παρέχεται από την πρόσδεση των υποδοχέων των Τ λεμφοκυττάρων (ΤCR) με τα αντιγόνα τα οποία φέρονται στα MHC (I και ΙΙ) μόρια των APC, και ένα συν-διεγερτικό σήμα. Υπεύθυνες γι’αυτό είναι πρωτεΐνες επιφανείας των APC, με κυριότερες εκπροσώπους τις B7.1 (CD80) και B7.2 (CD70). Οι B7.1 διεγείρουν την παραγωγή των Τh1 λεμφοκυττάρων, ενώ οι Β7.2 των Th2 (17) (Εικόνα 1).
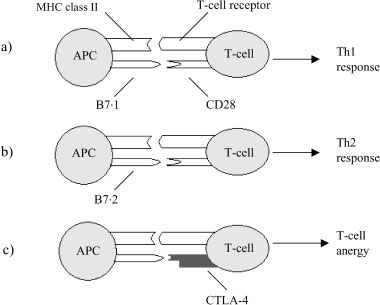
Εικόνα 1. Εναλλακτικό αποτέλεσμα της αντιγονοπαρουσίασης ανάλογα με το συνδιεγερτικό μόριο: (a) Σύνδεση του B7·1 με το CD28 επάγει την διέγερση Th1 λεμφοκυττάρων (b) Σύνδεση του B7·2 με το CD28 επάγει την διέγερση Th2 λεμφοκυττάρων (c) Αλληλεπίδραση του CTLA-4 με το B7·1 ή B7·2 αναστέλλει τη διέγερση των T λεμφοκυττάρων οδηγώντας σε ανέργια. Fountoulakis S & Tsatsoulis A, Clin Endocrinol (Oxf) 2004,with permission.
Η αλληλεπίδραση των Β7 πρωτεϊνών με το CD28 αντιγόνο επιφανείας των ανενεργών Τ λεμφοκυττάρων οδηγεί στην ενεργοποίησή τους, ενώ πρόσδεση στο CTLA-4 αντιγόνο επιφανείας ήδη ενεργοποιημένων Τ λεμφοκυττάρων έχει ως αποτέλεσμα τη μετάδοση αρνητικού σήματος. To CTLA-4 απαντάται και σε διαλυτή μορφή (s-CTLA-4) με άγνωστο μέχρι στιγμής ρόλο (18). Δύο νέα ζεύγη υποδοχέων-συνδετών έχουν εμπλακεί στη μετάδοση του σήματος ενεργοποίησης των λεμφοκυττάρων. Η Β7RP-1 (ICOSL) πρωτεΐνη προσδένεται στον υποδοχέα ICOS και μεταδίδει διεγερτικό σήμα (19), ενώ η πρόσδεση των B7-H1 (PD-L1) και B7-DC (PD-L2) με τον υποδοχέα PD-1 μεταδίδει ανασταλτικό σήμα (20).
Σημαντικό ρόλο στην έκβαση της αντιγονοπαρουσίασης και της ανοσολογικής απάντηση φαίνεται πως έχουν και κάποιες σχετικά νέες κατηγορίες λεμφοκυττάρων (T ρυθμιστικά κύτταρα /Τ regulatory cells/Tregs, Τh17, Τh22 και Tfh/Τ follicular helper cells) αλλά και τα Β λεμφοκύτταρα.
- Tα Τregs αποτελούν μια ομάδα CD4 λεμφοκυττάρων τα οποία εκφράζουν έντονα το αντιγόνο επιφανείας CD25 (IL2a-receptor) και τον ενδοκυττάριο μεταγραφικό παράγοντα FoxP3. Έχουν κύριο ρόλο στη ρύθμιση της ανοσολογικής απάντησης ασκώντας κατασταλτική δράση στα CD4 και CD8 λεμφοκύτταρα, συμπεριλαμβανομένων των αυτοαντιδραστικών λεμφοκυττάρων που διαφεύγουν της κεντρικής ανοχής ή εμφανίζονται de novo (Τ effectors). Τα τελευταία έτη έχουν περιγραφεί πολλές διαφορετικές υποομάδες των Tregs (induced-iTregs, Τr1, natural occurring-nTregs) χωρίς ακόμα να είναι σαφής ο βαθμός κατασταλτικής δράσης καθεμιάς (21,22). Για την ενεργοποίησή τους είναι απαραίτητη η αλληλεπίδραση με τα APC και τα CD4 λεμφοκύτταρα μέσω των αντιγόνων επιφανείας τους. Για την επιβίωσή τους είναι απαραίτητη η IL-2 ενώ η δράση τους ασκείται μέσω IL-10 που παράγουν και η οποία δρα στη συνέχεια στα Τ λεμφοκύτταρα. Ρόλος τους είναι η αποφυγή της ανάπτυξης οργανοειδικής αυτοανοσίας. Τα Τregs ασκούν τη δράση τους μέσω άμεσης επαφής ή/και μέσω παραγωγής κυτοκινών. Για να είναι λειτουργικά πρέπει να ενεργοποιηθούν μέσω του TCR υποδοχέα οπότε πολλοί ερευνητές δέχονται πως για την κατασταλτική τους δράση είναι απαραίτητη η ταυτόχρονη αλληλεπίδραση Tregs-APC-CD4 λεμφοκυττάρων (23) με τα APC να έχουν ρόλο σταθεροποιητικής πλατφόρμας.
- Τα Th17 λεμφοκύτταρα αποτελούν μια προσφάτως ανακαλυφθείσα υποκατηγορία CD4 λεμφοκυττάρων που παράγουν IL-17 και ΙL-22 (24) και φέρουν τα αντιγόνα επιφανείας: T-cell Receptor (TCR), CD3, CD4 και CCR6. Ο μεταγραφικός παράγοντας RORγt (retinoid related orphan receptor) έχει σημαντικό ρόλο στον έλεγχο της διαφοροποίησής τους ενώ η ΙL-23, που παράγεται από APC, είναι απαραίτητη για τη λειτουργία τους ευοδώνοντας την παραγωγή άλλων κυτοκινών και αυξάνοντας τον RORγt (25). Ειδικότερα, οι παράγοντες TGF-b και IL-6 ενεργοποιούν τον RORγt που βρίσκεται στα CD4 λεμφοκύτταρα και τα διαφοροποιούν προς την Τh17 κατεύθυνση (26). Τα Τh 17 λεμφοκύτταρα έχουν ρυθμιστικό ρόλο στην κινητοποίηση, επιστράτευση και ενεργοποίηση των ουδετερόφιλων ενώ μπορούν να επάγουν την έκκριση κυτοκινών από άλλα κύτταρα (π.χ. APC), έχοντας κυρίως προφλεγμονώδη δράση. Η IL-22 έχει και αυτή προφλεγμονώδη δράση αν και έχει αναφερθεί και προστατευτική δράση στα επιθηλιακά κύτταρα (27-30).
- Τα Τh22 λεμφοκύτταρα αποτελούν ακόμα μια υποομάδα των T λεμφοκυττάρων που φέρουν τα αντιγόνα TCR, CD3, CD4, CCR6, CCR4 και CCR10 και παράγουν IL-22 αλλά όχι IL-17. Η IL-22 έχει προφλεγμονώδη δράση σε συνέργεια με την IL-17, τον TNF-α και την IFN-γ αλλά μπορεί να παίξει και προστατευτικό ρόλο στην επιβίωση των επιθηλιακών κυττάρων (27-30).
- Tα Τfh λεμφοκύτταρα συνιστούν ένα προσφάτως περιγραφέν υποείδος Τ λεμφοκυττάρων (CD4+CXCR5+ICOShigh ή CD4+CXCR5+PD-1high) και η διαφοροποίηση τους συντελείται με τη συμβολή του μεταγραφικού παράγοντα Βcl-6 τον οποίο εκφράζουν (31). Τα Τfh λεμφοκύτταρα δύναται να μεταναστεύουν σε λεμφοειδή κέντρα (germinal centers) μέσω των υποδοχέων που φέρουν (CXCR5), η έκφραση των οποίων είναι απαραίτητη τόσο για τη μετανάστευση όσο και τη λειτουργία τους. Συμμετέχουν στη ρύθμιση της αντιγονοειδικής ανοσίας και της ανοσολογικής απάντησης των Β λεμφοκυττάρων και την παραγωγή αντισωμάτων (32) μέσω κυτοκινών που παράγουν (κυρίως IL-21) καθώς και των αντιγόνων επιφανείας που φέρουν.
2.2. Μηχανισμοί ανοσολογικής ανοχής και διαφυγής από αυτήν.
Εδώ περιλαμβάνονται μηχανισμοί που προστατεύουν τον οργανισμό από την εμφάνιση λεμφοκυττάρων (T effectors) αντιδραστικών έναντι αντιγόνων του ίδιου του οργανισμού (αυτοαντιγόνα). Διαταραχή σε έναν ή περισσότερους από αυτούς τους μηχανισμούς μπορεί να οδηγήσει στην εμφάνιση αυτοανοσίας. Τέτοιοι μηχανισμοί είναι η κεντρική (στο θύμο) και περιφερική ανοχή (central and peripheral tolerance) καθώς και η ανέργια των αυτοαντιδραστικών κλώνων των λεμφοκυττάρων (clonal anergy). Οι πρώτες δύο συντελούν στην εξάλειψη των ανώριμων αυτοαντιδραστικών κλώνων, λόγω απουσίας συν-διεγερτικού σήματος (πχ. Β7.1 ή Β7.2 με CD28) μετά από επαφή με αυτοαντιγόνα. Η ανέργια επιτυγχάνεται όταν ώριμα ανοσολογικά κύτταρα έρχονται σε επαφή με αυτοαντιγόνα απουσία συν-διεγερτικού σήματος και συμβάλλει στην υπορρύθμιση και λειτουργική ανενεργοποίησή τους (33). Σε πρώτο στάδιο τα εν δυνάμει αυτοαντιδραστικά Τ λεμφοκύτταρα καταστρέφονται στο θύμο μέσω των μηχανισμών της κεντρικής ανοχής. Στο δεύτερο στάδιο τα λίγα κύτταρα που πιθανώς διέφυγαν της κεντρικής ανοχής και μετανάστευσαν στην περιφέρεια ελέγχονται μέσω των μηχανισμών της περιφερικής ανοχής (ανέργια, καταστολή από τα Tregs).
Η απομόνωση του γονιδίου AIRE (autoimmune regulator), υπεύθυνου για το αυτοάνοσο πολυαδενικό σύνδρομο τύπου 1, έριξε νέο φως στη θεώρηση της κεντρικής ανοχής ως σημαντικού ρυθμιστικού μηχανισμού της αυτοανοσίας. Η πρωτεΐνη ΑΙRΕ εκφράζεται κυρίως σε επιθηλιακά κύτταρα του μυελού του θύμου και σε μονοκύτταρα-δενδριτικά κύτταρα που εντοπίζονται σε αυτόν. Μελέτες σε ποντίκια στα οποία απουσιάζει το ΑΙRE έδειξαν πλήρη ανικανότητα εξάλειψης ορισμένων οργανοειδικών κυττάρων στο θύμο. Η ανακάλυψη πως η θυρεοσφαιρίνη μπορεί να εκφράζεται στο μυελό του θύμου, οδήγησε στην υπόθεση πως η συνεισφορά του AIRE στην ανοχή των θυρεοειδικών αυτοαντιγόνων ίσως είναι σημαντική. Μειωμένη έκφραση του AIRE ίσως οδηγεί σε μείωση της έκφρασης της Tg και άλλων θυρεοειδικών αυτοαντιγόνων στο θύμο που τελικά οδηγεί στη διαφυγή αυτοαντιδραστικών Τ λεμφοκυττάρων στην περιφέρεια (34,35).
Από την άλλη πλευρά και η περιφερική ανοχή συμβάλλει στην προστασία από την εμφάνιση αυτοαντιγόνων. Μπορεί να επιτευχθεί μέσω της αλληλεπίδρασης των APC με αυτοαντιδραστικά Τ λεμφοκύτταρα που φέρουν το κατασταλτικό μόριο CTLA-4. Ακόμα τόσο η διαλυτή μορφή sCTLA-4, όσο και τα νέα μόρια PD-1 και PD-L1, PD-L2 φαίνεται πως διαδραματίζουν σπουδαίο ρόλο στον περιορισμό πιθανών αυτοαντιδραστικών κυττάρων(18,19,20). Πρέπει βέβαια να σημειωθεί πως η συνεισφορά των παραπάνω, προσφάτως ανακαλυφθέντων, μορίων στην περιφερική ανοχή δεν έχει πλήρως διευκρινιστεί και χρειάζεται περαιτέρω μελέτη.
3. Αιτιοπαθογένεια θυρεοειδικής αυτοανοσίας
3.1. Γενετικοί παράγοντες
Η αλληλεπίδραση περιβαλλοντικών παραγόντων με το γενετικό υπόβαθρο ορισμένων ατόμων διεκδικεί κύριο ρόλο στην παθογένεια της ΘΑ. Όσον αφορά στη γενετική προδιάθεση για ανάπτυξη ΘΑ, έχουν εμπλακεί γονίδια του μείζονος συμπλέγματος ιστοσυμβατότητας (MHC), που θεωρούνται και τα πιο σημαντικά, αλλά και μη-MHC γονίδια. Μεγάλος αριθμός μελετών έχει συσχετίσει διαφορετικούς εθνολογικά αλλήλους του MHC γονιδίου με αυξημένη πιθανότητα εμφάνισης HT και GD. Ακόμα, πολυμορφισμοί γονιδίων όπως τα CTLA-4, FCRL3, CD40, PTPN22, CD25, TSHR, Tg, SCGB3A2 και γονιδίων που κωδικοποιούν προφλεγμονώδεις κυτταροκίνες φαίνεται να συνδράμουν στην εμφάνιση της ΘΑ.
Η σημασία του γενετικού υποβάθρου στην εμφάνιση της ΘΑ διαπιστώθηκε αρχικά από μελέτες διδύμων οι οποίες κατέγραψαν μεγαλύτερο ποσοστό ταυτόχρονης παρουσίας (concordance) της νόσου και στα δύο αδέλφια σε μονοζυγωτικούς σε σχέση με τους διζυγωτικούς διδύμους. Ωστόσο ακόμα και στους μονοζυγωτικούς διδύμους το ποσοστό αυτό δεν ξεπερνούσε το 50% (36) παρέχοντας ενδείξεις για τη συμμέτοχη και περιβαλλοντικών παραγόντων. Περαιτέρω μελέτες σε διδύμους έδειξαν πως περίπου 80% του κινδύνου για ανάπτυξη ΘΑ οφείλεται σε γενετικούς παράγοντες ενώ οι περιβαλλοντικοί και λοιποί παράγοντες συνεισφέρουν στο 20% (36,37).
Σε δύο ενδιαφέρουσες ανασκοπήσεις του Tomer και των Brand & Gough (38,39) oι μελέτες οι οποίες στοχεύουν στην αναγνώριση και χαρτογράφηση γονιδίων με συνεισφορά στην προδιάθεση ανάπτυξης ΘΑ, κατατάσσονται χρονολογικά στις εξής κατηγορίες.
- Πρώιμες μελέτες ανάλυσης υποψήφιων γονιδίων (candidate gene analysis). Στα υποψήφια γονίδια κατατάσσονται αυτά που, λόγω της λειτουργίας τους, θεωρούνται πως μπορεί να εμπλέκονται στην αιτιοπαθογένεια της ΘΑ (π.χ τα γονίδια που κωδικοποιούν την ΤPO, Tg, NIS ή τον υποδοχέα της TSH -ΤSHR).
- Μελέτες ελέγχου σύνδεσης σε επίπεδο γονιδιώματος (whole–genome linkage screening). Πρόκειται για μελέτες ελέγχου ολόκληρου του γονιδιώματος για κάθε πιθανό γονίδιο που εμπλέκεται στην αιτιοπαθογένεια της νόσου. Στις μελέτες αυτές επιστρατεύονται οικογένειες στις οποίες η νόσος ενδημεί. Τα μέλη που νοσούν ελέγχονται με δείκτες γονοτύπισης (genotyping markers) όλου του γονιδιώματος για πολυμορφισμούς ενός νουκλεοτιδίου (SNPs/single nucleotide polymorphisms) ή μικροδορυφόρους (microsatellites). Οι τελευταίοι αποτελούν επαναλήψεις δινουκλεοτιδίων 1-6 ζευγών βάσεων. Τετρακόσιοι μικροδορυφορικοί δείκτες είναι αρκετοί για τον έλεγχο όλου του γονιδιώματος μια και οι μικροδορυφόροι δεν απαντώνται σε μεγάλη γειτνίαση μεταξύ τους, σε αντίθεση με τους SNPs που είναι πολύ συχνοί (περίπου ένας κάθε 300 ζεύγη βάσεων) οπότε για τον έλεγχο όλου του γονιδιώματος απαιτούνται πολύ περισσότεροι SNP δείκτες.
Βασικό εργαλείο των μελετών αυτών αποτελεί η ανάλυση σύνδεσης (linkage analysis) η οποία στηρίζεται στο ότι η πιθανότητα ανασυνδυασμού δύο γονιδίων μειώνεται όσο πιο κοντά βρίσκονται το ένα με το άλλο. Εφόσον υπάρξουν στοιχεία σύνδεσης των SNPs που απαντώνται σε μια συγκεκριμένη περιοχή (locus) του γονιδιώματος με τη μελετώμενη νόσο, τότε η μελέτη συνεχίζεται με λεπτομερή χαρτογράφηση (fine mapping) της περιοχής αυτής. Στους περιορισμούς των μελετών αυτών περιλαμβάνονται η ύπαρξη γενετικών παραλλαγών, ο μικρός συνήθως αριθμός ατόμων και η έλλειψη λεπτομερούς μεθοδολογίας γονοτύπισης. Η δημιουργία λεπτομερών χαρτών (linkage maps) των μικροδορυφορικών δεικτών έδωσε νέα πνοή σε αυτού του είδους τις μελέτες.
- Μελέτες συσχέτισης σε επίπεδο γονιδιώματος (genome–wide association studies–GWAS). Τα τελευταία έτη οι GWAS αποτελούν ένα πολύ ευαίσθητο εργαλείο μια και μπορούν να ανιχνεύσουν γονίδια τα οποία συμβάλλουν ακόμα και σε ποσοστό μικρότερο του 5% στη γενετική προδιάθεση μιας νόσου. Αρχή των μελετών αυτών είναι η σύγκριση της συχνότητας ενός αλληλίου π.χ. ΗLA-DR3 στους ασθενείς σε σχέση με μια ομάδα ελέγχου. Εάν το αλλήλιο συσχετίζεται με τη νόσο θα εμφανίζεται πιο συχνά στους ασθενείς. Οι GWAS έγιναν εφικτές μετά την ολοκλήρωση του HapMap Project με το οποίο επιτεύχθηκε η γονοτύπιση 3,5 εκ. SNPs και περιγράφηκε η μεταξύ τους ανισορροπία σύνδεσης (LD-linkage disequilibrium). H σημασία της LD στις GWAS μελέτες είναι μεγάλη, μια και το μελετούμενο γονίδιο ίσως δεν έχει, αυτό καθεαυτό, ρόλο στην προδιάθεση για τη νόσο, όσο η σύνδεσή του με κάποιο άλλο γονίδιο. Έτσι ενώ μέχρι την εποχή των GWAS χρησιμοποιούνταν ξεχωριστοί δείκτες για κάθε έναν SNPs, πλέον μπορούν να χρησιμοποιούνται tag-SNPs δείκτες που ο καθένας αφορά σε μια ευρύτερη περιοχή LD (LD block). Έχει βρεθεί πως τα γονίδια που βρίσκονται σε μια περιοχή LD σπάνια ανασυνδυάζονται μεταξύ τους, ενώ ο ανασυνδυασμός είναι πιο πιθανός στην περιοχές μεταξύ των LD. Έτσι είναι εφικτός ο έλεγχος ολόκληρου του γονιδιώματος με τη χρήση 500000 τέτοιων SNPs. Έως τώρα τέτοιου εύρους έλεγχος δεν έχει γίνει για τη ΘΑ. Ωστόσο πολύ πρόσφατα η μελέτη WTCCC χρησιμοποίησε 14500 SNPs ενώ το Ιmmune chip Project περιλαμβάνει 200000 SNPs (40,41,42).
- Αλληλούχιση σε επίπεδο γονιδιώματος (whole–genome sequencing). Το επόμενο βήμα στις γονιδιακές μελέτες είναι η δημιουργία έτοιμων πλακετών (platforms) αλληλούχισης ολόκληρου του ανθρώπινου γονιδιώματος. Η ανάλυση των δεδομένων που θα προκύψουν από τέτοιες μελέτες μπορεί να παρέχει πληροφορίες για το γενετικό κίνδυνο εμφάνισης μιας νόσου σε κάθε άτομο ξεχωριστά. Επίσης η εφαρμογή της τεχνολογίας ChIP-Seq (ανοσοκαθήλωση χρωματίνης σε επίπεδο γονιδιώματος και αλληλούχιση) μπορεί να παρέχει λεπτομερή χαρτογράφηση όσον αφορά στην πρόσδεση μεταγραφικών παραγόντων στο γενετικό υλικό και την επακόλουθη ρύθμιση της μεταγραφής του.
Τα γονίδια, οι πολυμορφισμοί των οποίων έχουν ενοχοποιηθεί για την εμφάνιση προδιάθεσης για ΘΑ, μπορούν να χωριστούν σε πέντε κατηγορίες.
- Γονίδια που εμπλέκονται στην αλληλεπίδραση των Τ και Β λεμφοκυττάρων με τα ΑPC ή αλλιώς γονίδια ανοσολογικής σύναψης (immunological synapse genes): ΗLA-DR, CTLA-4, FCRL3, CD40, ΑΙRE, PTPN22, TCR-β
- Γονίδια ρυθμιστικών Τ λεμφοκυττάρων (Τregs): CD25(IL2aR), FOXP3
- Γονίδια πρωτεϊνών του θυρεοειδούς: Tg, TSHR, ΤPO
- Γονίδια κυτοκινών και υποδοχέων τους: IL23R, TNF-a, IL-13, IL-3, IL-4, IL-6, IL-17F, IL-1RN
- Λοιπά γονίδια που προέκυψαν από GWAS: SCGB3A2, ΜΜΕL1, LPP, BACH2, FGFR1oP, PRICKLE1, 216 kb upstream of TRIB2, 83 kb upstream of ITGAM, CCR6. Επίτοποι στα χρωμοσώματα Χq21.1, 22q12.3-13.1, 1q23.2, 9q34.2, 14q32.2, 10q rs6479778, 14q rs12147587, rs2284720.
Για κάποια από τα παραπάνω γονίδια υπάρχουν λεπτομερή και ισχυρά στοιχεία για τη συμβολή τους στην προδιάθεση για ΘΑ, από πολλές και νέας μεθοδολογίας μελέτες, ενώ για άλλα απαιτούνται μεγαλύτερες και πιο ακριβείς μελέτες.
3.1.1. Γονίδια των οποίων η εμπλοκή στην προδιάθεση για ΘΑ έχει επιβεβαιωθεί από GWAS μελέτες
ΗLA–DR (6p21). Τα γονίδια HLA-I και ΙΙ κωδικοποιούν ετεροδιμερείς πρωτεΐνες (ΜΗC-I και MHC-II) υπεύθυνες για την αντιγονοπαρουσίαση. To γονίδιο ΗLA-DR3 (ΗLA-II) αποτελεί ένα από τα πρώτα γονίδια που μελετήθηκαν και εμφανίζει ισχυρή συσχέτιση με την GD (43). Ορισμένα αλλήλιά του, επιτρέπουν σε αυτοαντιγονικά πεπτίδια να ταιριάζουν καλύτερα στις θέσεις πρόσδεσης του MHC-II (peptide binding pockets) και έτσι η παρουσίαση τους στα CD4 λεμφοκύτταρα να είναι πιο αποτελεσματική. Πρόσφατες μελέτες έδειξαν πως η ύπαρξη Αrg στη θέση 74 της αλυσίδας HLA-DRb1 (peptide binding pocket) οδηγεί σε προδιάθεση για την εμφάνιση GD (44), ενώ αντίστοιχο αμινοξύ έχει βρεθεί και για την HT (45). Εκτός από γονίδια σε περιοχές του ΗLA-II (HLA-DRB1 και HLA-DQA1), στην αιτιοπαθογένεια της ΘΑ έχουν εμπλακεί και γονίδια στην περιοχή ΗLA I (HLA-C, HLA-B) (44,46). Όπως προαναφέρθηκε, οι ΗLA-I πρωτεΐνες επιφανείας (ΜΗC-I) προσδένουν βακτηριακά/ιικά πεπτίδια και τα παρουσιάζουν στα κυτταροτοξικά CD8 κύτταρα.
CTLA-4 (2q33). Πολυμορφισμοί του CTLA-4 γονιδίου oι οποίοι μειώνουν την έκφραση ή/και λειτουργία του, οδηγούν στην παραγωγή αυτοαντιδραστικών Τ λεμφοκυττάρων και συσχετίζονται με την εμφάνιση ΗΤ και GD (47-53). Η συμμετοχή του CTLA-4 στην προδιάθεση για ΘΑ είναι σύνθετη μια και τόσο το προστατευτικό αλλήλιο Α όσο και το προδιαθεσικό G μπορούν να ευθύνονται, αν αλληλεπιδράσουν με άλλους επιτόπους (loci). Δεν είναι ακόμα γνωστό ποια παραλλαγή (variant) έχει τη μεγαλύτερη συμμετοχή. Έως τώρα, τέσσερεις πολυμορφισμοί έχουν μελετηθεί με λεπτομερή χαρτογράφηση: ΑΤ μικροδορυφόρος στην 3’UTR περιοχή, A/G SNP θέσης 49, A/G SNP έξω από την 3’ UTR περιοχή και non-coding intron 1 παραλλαγή (47-53) με τον πρώτο και τον τρίτο να αποτελούν τους πιθανότερους υποψήφιους.
FCRL3 (1q22). Αποτελεί μέλος της υπεροικογένειας των υποδοχέων ανοσοσφαιρινών. Η πρωτεΐνη που κωδικοποιεί το γονίδιο FCRL3 περιέχει ενεργοποιητικά και κατασταλτικά μοτίβα στην κυτταροπλασματική της περιοχή (immunoreceptor-tyrosine activation and inhibitory motifs) και ελέγχει παραμέτρους της μετάδοσης σήματος των Β κυττάρων. Πολυμορφισμοί του γονιδίου έχουν ενοχοποιηθεί για προδιάθεση ανάπτυξης ΘΑ (42,54).
CD40 (20q13). Το CD40 εκφράζεται κυρίως στα Β λεμφοκύτταρα και APC και παίζει βασικό ρόλο στην ενεργοποίησή τους και την έκκριση αντισωμάτων. Τα θυρεοκύτταρα μπορούν επίσης να το εκφράσουν στην επιφάνειά τους (55). Έχει βρεθεί πως η σύνδεση CD40/CD40L επάγει την εμφάνιση και αύξηση των CD8 Tregs. Μελέτες σύνδεσης σε επίπεδο γονιδιώματος με λεπτομερή χαρτογράφηση (whole-genome linkage studies) ανέδειξαν έναν επίτοπο του CD40 στο χρωμόσωμα 20q ο οποίος συσχετίζεται με προδιάθεση για GD (56-58) . Ο επίτοπος αυτός αποτελεί έναν C/T SNP πολυμορφισμό στην αλληλουχία Kozak που αυξάνει την έκφραση του CD40 στα Β, τα ΑPC ή/και τα θυρεοκύτταρα (59) και οδηγεί σε αυξημένη παραγωγή anti-TSHR αντισωμάτων.
PTPN22 (protein tyrosine phosphatase 22) (1q13). Το γονίδιο αυτό κωδικοποιεί τη λεμφοειδική τυροσινική φωσφατάση (lymphoid tyrosine phosphatase) η οποία είναι ανασταλτικός ρυθμιστής της ενεργοποίησης των Τ λεμφοκυττάρων (60). Ο εμπλεκόμενος SNP οδηγεί στην αντικατάσταση της αργινίνης με τρυπτοφάνη και τη δημιουργία μιας ενεργοποιητικής (gain of function) παραλλαγής στην θέση 620 (R620W) (61,62). Συσχετίζεται τόσο με την προδιάθεση για ανάπτυξη GD όσο και με ΗΤ λόγω της επακόλουθης μειωμένης ενεργοποίησης των Τ κυττάρων που ίσως επιτρέπει σε αυτοαντιδραστικά Τ λεμφοκύτταρα (T effectors) να διαφύγουν των μηχανισμών της θυμικής ανοχής.
ΙL2Ra/ CD25 (10q15). Η ύπαρξη πολυμορφισμών του CD25 έχει συσχετιστεί με την εμφάνιση προδιάθεσης για GD (63). Οι πολυμορφισμοί αυτοί μάλλον εμπλέκονται στην κατασταλτική λειτουργία των Τregs και όχι τόσο στη μεταβολή του αριθμού τους, μια και τα επίπεδά των Tregs δεν έχει αποδειχτεί με βεβαιότητα πως σχετίζονται με την εμφάνιση ΘΑ (64).
TSHR (14q13). Οι μελετώμενοι πολυμορφισμοί αφορούν σε περιοχές ιντρονίων που δεν κωδικοποιούν πρωτεΐνες (non-coding intronic SNPs) (65,66,67). Ο πιο σημαντικός από αυτούς ( intron 1 SNP) μπορεί να μεταβάλλει το εναλλακτικό μάτισμα (splicing) του TSHR (68). Η εμπλοκή όλων των παραλλαγών (variants) που εντοπίζονται μέσα και αμέσως έξω από την 40 Κp περιοχή του ιντρονίου 1 καθώς και του ιντρονίου 7 απαιτεί περαιτέρω έρευνα.
SCGB3A2/ secretoglobin family 3A member 2 (5q32). Αρχικές μελέτες σύνδεσης (linkage studies) εντόπισαν την περιοχή 5q31-33 στην οποία περιέχονται πολλά υποψήφια γονίδια (π.χ IL13, IL4) προδιάθεσης για ΘΑ (69). Μελέτες συσχέτισης (association studies) ανέδειξαν το SCGΒ3Α2 ως σχετιζόμενο με προδιάθεση εμφάνισης ΘΑ (70). Ο πιο σημαντικός SNP είναι ο rs1368408 που συσχετίζεται με τη GD (71). Ωστόσο μια πρόσφατη μελέτη συσχέτισης έδειξε πως δεν μπορεί να αποκλειστεί το γεγονός και άλλα γονίδια στην περιοχή αυτή (π.χ IL-3) να εμπλέκονται στην παθογένεια της ΘΑ (72) .
Tg (8q24). Μελέτες σύνδεσης σε επίπεδο γονιδιώματος (whole genome linkage studies) απεκάλυψαν τη συσχέτιση πολυμορφισμών του γονιδίου της θυρεοσφαιρίνης με την εμφάνιση GD (73,74). H αλληλούχιση που ακολούθησε, απεκάλυψε τρεις παραλλαγές (A734S, V1027M, W1999R) που ίσως μεταβάλλουν την αποδόμηση της Tg στα ενδοσώματα, οδηγώντας σε παθογενετικά Tg πεπτίδια με αυξημένη αντιγονικότητα (75). Τα πεπτίδια αυτά ίσως ευνοούν την εμφάνιση αυτοαντιδραστικών Τ και Β λεμφοκυττάρων.
3.1.2. Νέα γονίδια από GWAS με ισχυρά στοιχεία συσχέτισης για προδιάθεση ΘΑ
Επτά νέοι επίτοποι (loci) ανακαλύφθηκαν πρόσφατα στη Μ. Βρετανία με τη χρήση του WTCCC ImmunoChip (40-42): ΜΜΕL1, LPP, BACH2 (ανοσολογική απάντηση έναντι ιών), FGFR1oP, PRICKLE1, 216 kb upstream of TRIB2, 83 kb upstream of ITGAM, CCR6 (lymphocyte trafficking) (76,77). Πρόσφατα μια ομάδα ερευνητών από την Κίνα περιέγραψε ακόμα 5 νέους επιτόπους που συσχετίζονται με τη GD (Χq21.1, 22q12.3-13.1, 1q23.2, 9q34.2 14q32.2) (78). Επίσης λεπτομερής χαρτογράφηση στα χρωμοσώματα 10q και 14q απεκάλυψε νέους επιτόπους προδιάθεσης για ΘΑ (10q /SNP rs6479778 στην περιοχή του γονιδίου ΑRID5B το οποίο συσχετίζεται και με ρευματοειδή αρθρίτιδα) και για GD (14q/ SNP rs12147587, rs2284720 στην περιοχή των γονιδίων ΝΡΧΝ3 και TSHR αντίστοιχα) (79).
3.1.3. Γονίδια των οποίων η εμπλοκή στην προδιάθεση για ΘΑ απαιτεί επιβεβαίωση από μεγαλύτερες και νέας μεθοδολογίας μελέτες
HLA-B*46, TPO, IL23R, TNF-a, IL-13, IL-3, IL-4, IL-6 (80), IL-17F (81), IL-1RN, VDR (82), ZFAT (83), LTA, FOXP3, TCR-b, PSMB8-9, DIO2, ADRB2, FCGRIIa, FIH, PDCD1, NLRP1(84), VEGF (85), DNMT1 GD (86), MTRR HT (86), TBX21 (87), HLX (87), ΑΙRE.
Τα περισσότερα γονίδια που συσχετίζονται με την εμφάνιση προδιάθεσης για ΘΑ αφορούν στη GD. Ωστόσο, με την εξαίρεση της παραλλαγής ΗLA-DRb1 Arg 74 η οποία έχει ΟR (odds ratio) >5 για την εμφάνιση GD, τα υπόλοιπα έχουν μικρή συμβολή με OR < 1,5-2. Φαίνεται λοιπόν πως για να αναπτύξει ένα άτομο τη νόσο πρέπει να κληρονομήσει πολλά διαφορετικά προδιαθεσικά γονίδια. Η πιθανότητα όμως συνύπαρξης σε ένα άτομο πολλών γονιδίων με μικρό OR, μειώνει σημαντικά τη συχνότητα της νόσου στο γενικό πληθυσμό μια και δεν είναι γενετικά εφικτό η ΘΑ να είναι μια τόσο συχνή νόσος με ταυτόχρονη συνύπαρξη πολλών διαφορετικών προδιαθεσικών γονιδίων σε κάθε ένα άτομο ξεχωριστά. Η υπόθεση που έχει διατυπωθεί από την ομάδα του Tomer είναι πως είτε τα προδιαθεσικά γονίδια αλληλεπιδρούν μεταξύ τους αυξάνοντας το συνολικό OR εμφάνισης της ΘΑ, είτε πως ορισμένα προδιαθεσικά γονίδια έχουν αυξημένο OR αλλά μόνο σε κάποια υποκατηγορία της ΗΤ ή GD ενώ στο σύνολο των ασθενών έχουν αισθητά μικρότερο OR. Πρέπει ωστόσο να τονιστεί πως οι γονιδιωματικές μελέτες δείχνουν ότι τα πιο ισχυρά αποδεικτικά στοιχεία για προδιαθεσικούς πολυμορφισμούς αφορούν στο HLA(DR3) και στο CTLA-4 γονίδιο.
3.2. Περιβαλλοντικοί παράγοντες
Όπως προαναφέρθηκε, η ύπαρξη μόνο γενετικών προδιαθεσικών παραγόντων δεν αρκεί για να αιτιολογήσει πλήρως την αιτιοπαθογένεια της ΘΑ. Τουλάχιστον 20% του κινδύνου εμφάνισης ΘΑ οφείλεται σε περιβαλλοντικούς παράγοντες. Μεταξύ των παραγόντων που έχουν ενοχοποιηθεί για την ανάπτυξη ΘΑ είναι το κάπνισμα (88), οι στεροειδικές ορμόνες, το τραύμα, το ιώδιο (89), το stress (90), η έλλειψη σεληνίου, διάφορα φάρμακα (π.χ. αμιωδαρόνη, ιντερφερόνη-α, antiCD52) και μικροβιακοί οργανισμοί (π.χ. Yersinia Enterocolitica) (91,92). Για τους τελευταίους θεωρείται πως τα αντισώματα έναντι κάποιων από αυτούς ίσως αλληλεπιδρούν και με πρωτεΐνες του ανθρώπου λόγω δομικής ομοιότητας (μοριακή μίμηση). Μεταβολές στην εντερική χλωρίδα έχουν επίσης ενοχοποιηθεί για την πρόκληση ανοσολογικών αντιδράσεων και την ανάπτυξη αυτοανοσίας, όπως στην περίπτωση της φλεγμονώδους νόσου του εντέρου, της σκλήρυνσης κατά πλάκας, του σακχαρώδους διαβήτη τύπου 1 (ΣΔ1) και της ρευματοειδούς αρθρίτιδας, χωρίς όμως ακόμα να υπάρχουν αρκετές μελέτες που αφορούν στη ΘΑ (92).
Οι σημαντικότεροι περιβαλλοντικοί παράγοντες που μπορούν να συμβάλλουν στην αιτιοπαθογένεια της ΘΑ είναι το ιώδιο και το έντονο stress (90) ενώ τα τελευταία χρόνια πολλοί ερευνητές μελετούν τις επιπτώσεις της έλλειψης σεληνίου και της συμπληρωματικής φαρμακευτικής χορήγησής του.
3.2.1. Ιώδιο
Πολλές επιδημιολογικές μελέτες όσο και μελέτες σε πειραματόζωα έχουν αναδείξει την υπερβολική πρόσληψη ιωδίου ως σημαντικό παράγοντα παθογένειας των αυτοάνοσων θυρεοειδικών νόσων (89,93). Αρκετοί μηχανισμοί έχουν προταθεί για τον τρόπο με τον οποίο το ιώδιο μπορεί να προάγει τις νόσους αυτές. Η σύνδεση του ιωδίου με τη θυρεοσφαιρίνη αυξάνει την αντιγονικότητά της, αυξάνοντας παράλληλα τη συγγένειά της με τον TCR και ενισχύοντας την παρουσίασή της από τα APC στα Τ λεμφοκύτταρα (94). Αυτό έχει ως αποτέλεσμα την ενεργοποίηση αυτοαντιδραστικών λεμφοκυττάρων έναντι της θυρεοσφαιρίνης. Επίσης η οξείδωση του ιωδίου από τη θυρεοειδική υπεροξειδάση (TPO) οδηγεί στην παραγωγή μεσολαβητών, οι οποίοι έχουν άμεση καταστρεπτική δράση έναντι των θυρεοκυττάρων, λόγω οξείδωσης των μεμβρανικών λιπιδίων και πρωτεϊνών τους (95). Τέλος το ιώδιο φαίνεται να έχει θετική επίδραση στην ωρίμανση των δενδριτικών κυττάρων, στην αύξηση του αριθμού των κυκλοφορούντων Τ λεμφοκυττάρων και την ενίσχυση της παραγωγής ανοσοσφαιρινών (2, 96). Σε πειραματικό μοντέλο ΗΤ η χορήγηση ιωδίου επάγει την έκφραση του αποπτωτικού μορίου TRAIL στην επιφάνεια τόσο των λεμφοκυττάρων όσο και των θυρεοκυττάρων ενώ την έκφραση του υποδοχέα DR5 μόνο σε θυρεοκύτταρα, καθιστώντας τα έτσι ευάλωτα σε απόπτωση μέσω αλληλεπίδρασης με το TRAIL (97). Από την άλλη πλευρά, η έκφραση TRAIL στα θυρεοκύτταρα μπορεί να καταστείλει την αυτοάνοση θυρεοειδίτιδα (98) και να αυξήσει τον αριθμό των Tregs (99).
3.2.2. Stress
Οι αρχικές μελέτες που συνέδεαν το stress με τη ΘΑ αφορούσαν ασθενείς με GD που ανέφεραν κάποιο έντονο στρεσογόνο γεγονός πριν την εμφάνιση της νόσου (100,101). Σημαντική ήταν η παρατήρηση πως κατά τη διάρκεια του πολέμου στη Σερβία (1992-1995) η συχνότητα της GD αυξήθηκε ενώ έως τότε παρέμενε σταθερή από το 1971 (102). Αρκετοί μηχανισμοί έχουν προταθεί για την επίδραση του stress στα κύτταρα που συμμετέχουν στην ανοσολογογική απάντηση της ΘΑ. Tα περισσότερα κύτταρα του ανοσολογικού συστήματος εκφράζουν υποδοχείς γλυκοκορτικοειδών και κατεχολαμινών. Τα γλυκοκορτικοειδή καταστέλλουν την παραγωγή IL-12 από τα APC και αυξάνουν την παραγωγή IL-4, IL-10 από τα Th2 λεμφοκύτταρα, ευοδώνοντας έτσι τη χυμική ανοσία (103). Επίσης, τα αυξημένα επίπεδα κατεχολαμινών, που παρατηρούνται στο stress, καταστέλλουν την παραγωγή IL-12 και TNF-a ενώ αυξάνουν την παραγωγή ΙFN-γ από τα APC. Επιπρόσθετα, τα διεγερμένα από κατεχολαμίνες δενδριτικά κύτταρα ευνοούν την παράγωγή IL-4 και IL-17 από τα Τ effectors ευοδώνοντας τη δημιουργία ενός Th2/Th17 μικροπεριβάλλοντος (104). Όλα τα παραπάνω έχουν ως συνέπεια τη δημιουργία συνθηκών που ευνοούν την Th2 ανοσολογική απάντηση, η οποία με τη σειρά της ευοδώνει την επικράτηση της χυμικής ανοσίας, την παραγωγή αυτοαντιδραστικών αντισωμάτων και την εμφάνιση GD.
Ακόμα, η κατάθλιψη και το stress ευνοούν την παραγωγή IL-6 από τα APC (105) ενώ η αύξηση του ΝF-kb (αντίστοιχη με αυτή που συμβαίνει και στο οξειδωτικό στρες) μπορεί μακροπρόθεσμα να οδηγήσει στην παραγωγή κυτοκινών που συμβάλλουν στην εμφάνιση της ΘΑ (106). Τέλος αξιοσημείωτη είναι και η κλινική παρατήρηση πως η χορήγηση βενζοδιαζεπινών μπορεί να μειώσει το ποσοστό υποτροπής της GD σε ορισμένους ασθενείς (107).
3.2.3. Σελήνιο
Ο θυρεοειδής αδένας χαρακτηρίζεται από τη μεγαλύτερη περιεκτικότητα του ιχνοστοιχείου σελήνιο στον ανθρώπινο οργανισμό, λόγω των σεληνοπρωτεινών που περιέχει (αποιωδινάσες 1-3). Ελάχιστες ποσότητες σεληνίου είναι αρκετές για την επαρκή λειτουργία των αποιωδινασών. Η χορήγηση σεληνίου οδήγησε σε μειωμένη απελευθέρωση IL-2, IFN-g, TNF-a από τα περιφερικά λεμφοκύτταρα ευθυρεοειδικών ασθενών με ΗΤ, η οποία συσχετίστηκε με μείωση του τίτλου αντι-TPO (108). Επίσης μια σημαντική ανακάλυψη είναι πως το σελήνιο αυξάνει τον αριθμό Tregs σε μοντέλο αυτοάνοσης θυρεοειδίτιδας NOD.H-2(h4) ποντικών (109). Μελέτες έχουν δείξει πως η χορήγηση σεληνίου μειώνει τα επίπεδα των θυρεοειδικών αυτοαντισωμάτων και βελτιώνει την υπερηχογραφική εικόνα του θυρεοειδούς σε ασθενείς με ΗΤ και έγκυες γυναίκες με θετικά anti-TPO αντισώματα (110-112) Επίσης υπάρχουν ενδείξεις πως το σελήνιο μειώνει την εμφάνιση θυρεοειδίτιδας της λοχείας και υποθυρεοειδισμού ενώ βοηθά στην ταχύτερη επίτευξη ευθυρεοειδισμού στη GD και φαίνεται πως έχει ευνοϊκή επίδραση στην ήπια οφθαλμοπάθεια. Ωστόσο παρά τις παρατηρήσεις αυτές, η επιβεβαίωση της επίδρασης του σεληνίου στη ΘΑ απαιτεί μεγαλύτερες, διπλές-τυφλές τυχαιοποιημένες (RCT) μελέτες και αυτός άλλωστε είναι και ο λόγος που η χορήγηση σεληνίου δε συμπεριλαμβάνεται στις κατευθυντήριες οδηγίες θεραπείας της ΘΑ.
3.3. Micro–RNA
Αποτελούν μια νέα κατηγορία κατασταλτικών ρυθμιστών της γονιδιακής έκφρασης. Πρόσφατη μελέτη έδειξε πως η έκφραση των miRNA 154*, 376b και 431* είναι μειωμένη σε περιφερικά μονοπύρηνα κύτταρα ασθενών με GD ενώ αυξάνεται μετά από θεραπεία με μεθιμαζόλη (113). Στα γονίδια-στόχους (target genes) των miRNA συμπεριλαμβάνονται χυμοκίνες, γονίδια που εμπλέκονται σε σηματοδοτικά μονοπάτια κυτοκινών και ανοσολογικής απάντησης, στο μονοπάτι των G πρωτεινών και στο Wnt σηματοδοτικό μονοπάτι. Όσον αφορά στο ρόλο τους στην αιτιοπαθογένεια της ΘΑ, έχει διατυπωθεί η υπόθεση πως η μείωσή τους μπορεί να ευοδώνει ανοσολογικές απαντήσεις που οδηγούν σε GD. Πολύ ενδιαφέρον είναι ότι δύο από αυτά τα mi-RNA είναι αστερόμορφα (starform) (154*, 431*) που μέχρι πρόσφατα θεωρούνταν ανενεργά μη κωδικοποιούντα (non-coding) μόρια RNA τα οποία αποδομούνται. Ωστόσο τελευταίες μελέτες δείχνουν πως ο ρόλος τους είναι πιο σημαντικός απ’ότι αρχικά πιστευόταν μια και φαίνεται πως έχουν την ικανότητα να επιδρούν στη γονιδιακή έκφραση πολλαπλών γονιδίων (114,115).
3.4. DAMPs (danger associated molecular patterns)
Μια πρόσφατη θεωρία (danger hypothesis) για την εμφάνιση της ΘΑ αναδεικνύει τη σημασία των DAMPs (danger associated molecular patterns). Τα DAMPs αποτελούν ενδογενή μόρια (τμήματα γενωμικού DNA, heat shock πρωτεΐνες, ουρικό οξύ, κολλαγόνο, υαλουρονικό οξύ) που παράγονται από θνήσκωντα κύτταρα. Ενδιαφέρον παρουσιάζει το γεγονός πως στα θυρεοκύτταρα εκφράζονται Toll-like υποδοχείς, οι οποίοι αναγνωρίζουν τα DAMPs. Οι κατατετμημένες διπλές έλικες του DNA δύνανται να εισέρχονται στο κυτταρόπλασμα των θυρεοκυττάρων όπου ανιχνεύονται από DNA-sensοrs (ιστόνη Η2Β) και στη συνέχεια να επάγουν την έκφραση ΜΗC-II, συνδιεγερτικών μορίων και ιντερφερόνης από τα θυρεοκύτταρα πυροδοτώντας την έναρξη ανοσολογικής απάντησης. Στο θυρεοκύτταρο μπορούν, με τον ίδιο μηχανισμό, να εισέρχονται και θυρεοειδικά αυτοαντιγόνα (π.χ TSH-R) από κατεστραμμένα γειτονικά θυρεοκύτταρα τα οποία στη συνέχεια παρουσιάζονται από το MHC-II των θυρεοκυττάρων στα CD4 λεμφοκύτταρα (116).
3.5. Μικροχιμαιρισμός
Ο όρος μικροχιμαιρισμός περιγράφει τη συνύπαρξη γενετικά διαφορετικών και διακριτών πληθυσμών κυττάρων που προέρχονται από δύο διαφορετικά άτομα. Σε κάποιες περιπτώσεις πρόκειται για μεταφορά μικρού αριθμού κυττάρων από ένα άτομο σε ένα άλλο π.χ. σε περιπτώσεις μετάγγισης αίματος ή μεταμόσχευσης οργάνου, ενώ πιο συχνά αφορά στη μεταφορά κυττάρων από το έμβρυο στην έγκυο γυναίκα μέσω του πλακούντα και αντίστροφα. Ο μικροχιμαιρισμός εμβρυικών κυττάρων αναφέρεται στην ανίχνευση εμβρυικών κυττάρων στο περιφερικό αίμα, τους λεμφαδένες ή τους ιστούς (και στο θυρεοειδή) της μητέρας, ενώ ο μικροχιμαιρισμός μητρικών κυττάρων αναφέρεται στην ανίχνευση μητρικών κυττάρων στο περιφερικό αίμα ή τους ιστούς του εμβρύου. Τα μικροχιμαιρικά κύτταρα μπορούν να επιβιώσουν για πολλές δεκαετίες ή ακόμα και καθ’όλη τη διάρκεια της ζωής ενός ατόμου (117). Έχει περιγραφεί και η περίπτωση τα εμβρυικά μικροχιμαιρικά κύτταρα που ανιχνεύονται στη μητέρα όχι μόνο να παραμένουν μετά τον τοκετό αλλά και να μεταφέρονται και στο επόμενο έμβρυο όταν η γυναίκα μείνει ξανά έγκυος (118).
Στα εμβρυικά μικροχιμαιρικά κύτταρα περιλαμβάνονται τροφοβλάστες, Τ και Β λεμφοκύτταρα, μονοκύτταρα/μακροφάγα και ΝΚ κύτταρα αλλά και αιμοποιητικά και μεσεγχυματικά αρχέγονα κύτταρα (stem cells) (118,119). Τα αρχέγονα αυτά κύτταρα μπορούν, σε ορισμένες περιπτώσεις, να μεταναστεύουν σε όργανα που έχουν υποστεί τραύμα ή βλάβη και να διαφοροποιούνται/ενεργοποιούνται εκεί, βοηθώντας στην αντιμετώπιση του βλαπτικού παράγοντα και των επιπτώσεών του (120,121). Ο ρόλος τους ωστόσο δεν είναι αποκλειστικά προστατευτικός μια και εμβρυικά μικροχιμαιρικά ενεργοποιημένα Τ, ΝΚ λεμφοκύτταρα και μονοκύτταρα/μακροφάγα μπορεί να εμπλέκονται στην έναρξη αυτοάνοσων νοσημάτων μέσω των κυτοκινών που παράγουν (allo-autoimmunity) ή μπορεί να αναγνωρίζονται ως ‘’μερικώς’’ εξωγενή με αποτέλεσμα την έναρξη ανοσολογικής απάντησης (auto-alloimmunity).
Η ανάπτυξη ανοσολογικής ανοχής έναντι των μικροχιμαιρικών εμβρυικών κυττάρων μπορεί να οφείλεται στην παραμονή και ωρίμανσή τους στο θύμο της εγκύου όπου καταστρέφονται όσα κύτταρα μπορούν δυνητικά να στραφούν εναντίον μητρικών αντιγόνων. Άλλη υπόθεση είναι πως μητρικά αντιγόνα που περνούν από τη μητέρα στο έμβρυο έχουν ως αποτέλεσμα τη δημιουργία εμβρυικών Τregs τα οποία στη συνέχεια περνούν στη μητέρα όπου επηρεάζουν το μητρικό ανοσολογικό σύστημα ώστε να μην κινητοποιηθεί εναντίον των εμβρυικών μικροχιμαιρικών κυττάρων (122).
Ο πιθανός ρόλος του μικροχιμαιρισμού στην αιτιοπαθογένεια της ΘΑ έχει να κάνει με την αρχική παρατήρηση πως o αριθμός των μικροχιμαιρικών κυττάρων αυξάνει όσο προχωρά η κύηση γεγονός που συμπίπτει με τη βελτίωση της ΘΑ κατά την κύηση και την επανεμφάνισή της μετά τον τοκετό. Πιο σημαντική είναι η παρατήρηση πως σε θυρεοειδείς αδένες γυναικών με ΗΤ και GD έχει βρεθεί εντονότερη παρουσία μικροχιμαιρικών κυττάρων σε σχέση με τα υγιή άτομα (123). Ακόμα πιο ενδιαφέρον είναι πως τα προδιαθεσικά γονίδια HLA-DQA1*501-DQB1*0201 και DQB1*0301 απαντώνται πιο συχνά σε μητέρες και παιδιά θετικά για μικροχιμαιρισμό. Εξάλλου σε πειραματικά μοντέλα ποντικών με αυτοάνοση θυρεοειδίτιδα (ΕΑΤ), εμβρυικά ανοσολογικά κύτταρα (Τ λεμφοκύτταρα και δενδριτικά) βρέθηκαν να συσσωρεύονται στους θυρεοειδείς των μητέρων. Ωστόσο παρά τις ανωτέρω παρατηρήσεις μόνο μία case-control μελέτη αναγνώρισε τη μητρότητα (parity) ως πιθανό παράγοντα κινδύνου για εμφάνιση ΘΑ (124) ενώ άλλες μεγάλες επιδημιολογικές δε βρήκαν συσχέτιση κάτι που υποδηλώνει πως ο μικροχιμαιρισμός ίσως έχει οριακή συνεισφορά στην ανάπτυξη ΘΑ (125,126).
3.6. Αιτιοπαθογένεια IgG4 θυρεοειδίτιδας
Οι παθογενετικοί μηχανισμοί που οδηγούν στην IgG4 θυρεοειδίτιδα δεν έχουν ακόμα αποσαφηνιστεί. Σε ορισμένες ΙgG4-σχετιζόμενες νόσους έχει παρατηρηθεί επικράτηση της Th2 ανοσολογικής απάντησης και αυξημένη παραγωγή Th2 κυτοκινών όπως οι IL-4, IL-5, IL-10, IL-13. Επίσης ο αριθμός των Τregs είναι αυξημένος στους προσβεβλημένους ιστούς αλλά και το περιφερικό αίμα των ασθενών. Η αυξημένη παραγωγή IL-10 από τα Tregs και τα Τh2 λεμφοκύτταρα ίσως ευοδώνει την παραγωγή IgG-4 από τα Β λεμφοκύτταρα ενώ η αυξημένη παραγωγή TGF-β, που επίσης παρατηρείται, επάγει τη δημιουργία ινώδους ιστού.
4. Ο ρόλος των υποκατηγοριών των Τα και Β λεμφοκυττάρων στη ΘΑ
4.1. Τregs στη ΘΑ
Πειράματα σε ποντίκια ανθεκτικά σε ανάπτυξη αυτοάνοσης θυρεοειδίτιδας (B cell deficient NOD.H-2h4 mice) έδειξαν πως μετά από παροδική εξάλειψη των Tregs με τη χρήση anti-CD25, αυτά ανέπτυξαν θυρεοειδίτιδα (127). Επίσης, η εξάλειψη των Tregs είχε ως αποτέλεσμα το 30% των C57BL/6 ποντικών που είναι ανθεκτικά σε ανάπτυξη GD να γίνουν ευαίσθητα σε επαγωγή υπερθυρεοειδισμού ενώ ο χειρισμός αυτός αύξησε τη σοβαρότητα της νόσου και σε BALB/c ποντίκια που ήταν ευαίσθητα στην ανάπτυξη υπερθυρεοειδισμού (128). Οι Μorris και συν. έδειξαν πως τα Tregs μεταβάλλουν την επιρρέπεια σε πειραματική αυτοάνοση θυρεοειδίτιδα τόσο σε naïve όσο και σε ευαισθητοποιημένα έναντι της Tg (mTgtolerized) ποντίκια (129). Η χορήγηση του αυξητικού παράγοντα GM-CSF προλαμβάνει και καταστέλλει την πειραματική αυτοάνοση θυρεοειδίτιδα στα ποντίκια και συνοδεύεται από αύξηση των CD4+CD25+ Tregs (130). Η επαγωγή των δενδριτικών κυττάρων με τη χρήση GM-CSF ίσως ενεργοποιεί ειδικά έναντι της θυρεοσφαιρίνης CD4+CD25+ Tregs και καταστέλλει την αυτοάνοση θυρεοειδίτιδα. Τα ποντίκια στα οποία χορηγήθηκε GM-CSF είχαν αυξημένο ποσοστό CD4+CD25+ Tregs ενώ λεμφοκύτταρα από τα ποντίκια αυτά παρήγαγαν μεγάλες ποσότητες IL-10. Άρα η στροφή της ανοσολογικής απάντησης προς Th2 ανταπόκριση (με τη χρήση εκλεκτικής ενεργοποίησης των δενδριτικών κυττάρων με GM-CSF) ίσως έχει θεραπευτική εφαρμογή σε αυτοάνοσες νόσους όπου κυριαρχεί η Th1 απάντηση, όπως η ΗΤ (131).
Οι μελέτες που αφορούν στο ποσοστό των Tregs στο περιφερικό αίμα ασθενών με ΘΑ έχουν αντικρουόμενα αποτελέσματα. Η έλλειψη ομοφωνίας ίσως οφείλεται στο διαφορετικό τρόπο χαρακτηρισμού των κυττάρων αυτών στις παλαιότερες μελέτες, στο μικρό αριθμό των ασθενών, τις διαφορετικές υποκατηγορίες Tregs που μετρώνται ή στο διαφορετικό αριθμό των Tregs ανάλογα με το στάδιο της νόσου. Έτσι, κάποιες μελέτες δε βρίσκουν διαφορά στον αριθμό των Tregs στη ΘΑ ενώ άλλες αναφέρουν μειωμένα ή αυξημένα επίπεδα, αρνητική συσχέτιση με τα TSΑb στη GD και επιστροφή στο φυσιολογικό μετά θεραπεία της HT (64,132-135). Φαίνεται πάντως πως τα κυκλοφορούντα Tregs είναι δυσλειτουργικά και λιγότερο ικανά να αναστείλουν τον πολλαπλασιασμό των αυτοαντιδραστικών Τ λεμφοκυττάτων (T effectors) (134). Στην περίπτωση της GD η μείωση της ικανότητας αυτής ίσως οφείλεται στην παραγωγή IFN-a από δενδριτικά κύτταρα η οποία επάγει την απόπτωση των Tregs (136). Η αναστολή παραγωγής της IFN-a αποκατέστησε τη λειτουργία των Tregs και μείωσε τα Th2 λεμφοκύτταρα σε ασθενείς με GD (136) ενώ αντίστοιχο αποτέλεσμα είχε και η ενδοθυρεοειδική ένεση δεξαμεθαζόνης (137).
Τα δυσλειτουργικά Τregs ίσως ευοδώνουν την απώλεια ανοχής έναντι των θυρεοειδικών αντιγόνων και την επακόλουθη ανάδειξη anti-TSHR αντισωμάτων. Η βελτίωση της GD στην κύηση ίσως οφείλεται και στην υπερπαραγωγή Tregs. Πως όμως παρακάμπτεται η δράση των Tregs στην περίπτωση της ΘΑ; Τα αυτοαντιδραστικά Τ λεμφοκύτταρα ίσως καταφέρνουν να παρακάμπτουν την καταστολή από τα Tregs μέσω κυτταροκινών που παράγουν ενώ καθώς η φλεγμονή λύεται, η κατασταλτική δράση των Tregs επανέρχεται. Με το χρόνο όμως τα αυτοαντιδραστικά Τ κύτταρα ίσως γίνονται λιγότερο ευαίσθητα στην μέσω Tregs καταστολή ή τα Tregs εξαντλούνται λειτουργικά.
4.2. Th1, Th2, Τh17, Th22, Tfh (follicular helper T cells), B λεμφοκύτταρα στη ΘΑ
4.2.1. Τh1 και Τh2 λεμφοκύτταρα
Στην ΗΤ παρατηρείται αυξημένος λόγος Th1/Th2 κυτοκινών ενώ θεωρείται πως τα APC εκφράζουν κυρίως B7.1 με αποτέλεσμα να προσφέρουν στα διηθούντα το θυρεοειδή λεμφοκύτταρα συνδιεγερτικό σήμα για την παραγωγή κυρίως Th1 κυτοκινών (IFN-γ, TNF-α, IL-2) (138). Αυτό έχει ως συνέπεια την αύξηση της ευαισθησίας των θυρεοειδικών κυττάρων σε απόπτωση (139). Τα θυρεοειδικά κύτταρα μπορούν και αυτά να εκφράζουν τόσο Β7.1 όσο και Β7.2 μόρια με επακόλουθη διέγερση της Th1 ή Th2 αντίστοιχα ανταπόκρισης. Όσον όμως αφορά στη διαφορική έκφραση Β7.1 στη ΘΑ οι μελέτες εμφανίζουν αντικρουόμενα αποτελέσματα. Η επικρατούσα άποψη είναι πως στη ΗΤ τα θυρεοκύτταρα εμφανίζουν ισχυρή έκφραση Β7.1 (140). Η παραγωγή Th1 έναντι των Th2 κυτοκινών στην ΗΤ φαίνεται πως δεν περιορίζεται στα ενδοθυρεοειδικά λεμφοκύτταρα αλλά περιλαμβάνει και τα περιφερικά CD4 και CD8 T λεμφοκύτταρα (141). Αν αυτό ισχύει, η αλληλεπίδραση των θυρεοειδικών κυττάρων που φέρουν Β7.1, με Τ λεμφοκύτταρα που φέρουν CD28 θα μπορούσε να οδηγήσει στην επαγωγή της ενεργοποίησης Τ αυτοαντιδραστικών λεμφοκυττάρων και σε τέτοια ρύθμιση των προαποπτωτικών και αντιαποπτωτικών μορίων που να ευνοεί την επιβίωση των λεμφοκυττάρων.
Σημαντικός είναι και ο ρόλος της παρουσίασης αντιγόνων από τα APC στα CD8 λεμφοκύτταρα μέσω της αλληλεπίδρασης του MHC-I με τον TCR. Ειδικότερα, η αναστολή της αντιγονοπαρουσίασης αυτής με τη χρήση εκλεκτικού αναστολέα του ανοσοπρωτεοσώματος (ΟΝΧ0914) σε μοντέλο αυτοάνοσης θυρεοειδίτιδας ποντικών NOD-H2, μείωσε τη λεμφοκυτταρική διήθηση και τη διαφοροποίηση των Τ λεμφοκυττάρων προς Th1 και Th17 (142). Αντίθετα σε πειραματικό μοντέλο ποντικών με GD ο ίδιος αναστολέας δεν είχε αποτέλεσμα. Αξίζει να αναφερθεί πως ο ρόλος του ανοσοπροτεοσώματος είναι η αποδόμηση πρωτεϊνών μέσω κασπασών και θρυψίνης. Τα APC, καθώς και τα T και B λεμφοκύτταρα εκφράζουν μια παραλλαγή του πρωτεοσώματος το οποίο, μέσω της διάσπασης πρωτεϊνών, παράγει με πιο αποτελεσματικό τρόπο ΜHC-I restricted T cell επιτόπους. Αυτό είναι ιδιαίτερα σημαντικό όσον αφορά τη λειτουργία των ΑPC στη ΗΤ (142).
Αντίθετα στη GD παρατηρείται κυρίως Th2 ανταπόκριση ως αποτέλεσμα της B7.2 συνδιέγερσης των λεμφοκυττάρων και παραγωγή κυρίως Th2 κυτοκινών (IL-4, IL-5, IL-10) (143). Αυτό ευνοεί την επακόλουθη διέγερση της χυμικής ανοσίας και την παραγωγή αυτοαντισωμάτων από τα Β λεμφοκύτταρα. Φαίνεται πως στη GD, οι Th2 κυτοκίνες μπορούν να μεταβάλλουν και την έκφραση του MHC-II ενισχύοντας την παρουσίαση αυτοαντιγόνων (π.χ.TSH-R) από τα θυρεοκύτταρα στα λεμφοκύτταρα (2). Επίσης ο αυξημένος λόγος TNFa/sCD40L, που ανιχνεύεται σε ασθενείς με GD, σχετίζεται με μείωση της ενεργότητας της νόσου και μετάβαση από Th2 σε Τh1 ανταπόκριση (144). Η σημασία της ισορροπίας μεταξύ Th1 και Th2 ανταπόκρισης στη διαφορική φαινοτυπική έκφραση της θυρεοειδικής αυτοανοσίας ενισχύεται και από την μελέτη των Coles και συν. στην οποία το 1/3 των ασθενών με σκλήρυνση κατά πλάκας ανέπτυξαν GD μετά από θεραπεία με το anti-CD52 μονοκλωνικό αντίσωμα Campath 1-H που χρησιμοποιείται για να εξαλείψει τα Τ λεμφοκύτταρα και να καταστείλει την κυτταρική ανοσία (145). Μια πιθανή εξήγηση για αυτό είναι πως τα Τ λεμφοκύτταρα που εμφανίζονται μετά τη δράση του αντισώματος επάγουν κυρίως την Th2 ανταπόκριση οδηγώντας στην ανάπτυξη διεγερτικών θυρεοειδικών αυτοαντισωμάτων και GD. Ωστόσο, τελευταίες μελέτες έχουν αναδείξει την παρουσία και των Τh1 λεμφοκυττάρων στην GD μετά την ανακάλυψη πως ο υποδοχέας της TSH μπορεί, δρώντας ως αντιγόνο, να πυροδοτήσει είτε Th1 είτε Th2 ανταπόκριση ανάλογα με το είδος των ΑPC και το συνδιεγερτικό σήμα που αυτά παρέχουν (146).
4.2.2. Th17/Th22 λεμφοκύτταρα
Tα αυξημένα επίπεδα των Τh17 κυττάρων έχουν εμπλακεί στην αιτιοπαθογένεια της HT και της GD (133,138,147,148). Ενεργό ρόλο φαίνεται πως έχει η ΤSLP (thymic stromal lymphopoietin), μια Th2 κυτοκίνη που παράγεται από κοκκιοκύτταρα. Τα αυξημένα επίπεδά της προάγουν τη διαφοροποίηση των Th17 κυττάρων σε ασθενείς με GD (148). Επίσης, δενδριτικά κύτταρα, ενεργοποιημένα από την ΤSLP, μπορούν να ενεργοποιήσουν τα CD4 λεμφοκύτταρα προς την Th2 και την Τh17 κατεύθυνση μέσω παραγωγής IL-23 (149,150). Πολυμορφισμοί του γονιδίου που κωδικοποιεί την TSLP ίσως εμπλέκονται στην εμφάνιση GD (148).
Ένα άλλο μόριο που έχει σημαντικό ρόλο στη μετάδοση σήματος της IL-17 ενώ ταυτόχρονα αποτελεί και μόριο προσέλκυσης (chemoattractant) για τα Th17 λεμφοκύτταρα είναι ο προσδέτης χυμοκινών CCL20 (chemokine ligand 20). Ο CCL20 εκφράζεται από μακροφάγα και λευκοκύτταρα και αλληλεπιδρά με τον CCR6 υποδοχέα που βρίσκεται στα Th17 λεμφοκύτταρα. Τα επίπεδα πλάσματος του CCL20 είναι αυξημένα σε ασθενείς με GD και συσχετίζονται θετικά με τα επίπεδα της οστεοποντίνης (ΟPN) ενώ αυξημένα έχουν βρεθεί και τα επίπεδα αίματος των CXCL9 και CXCL11 σε ασθενείς με ενεργό GD (146, 151).
H OPN είναι μια πρωτείνη του εξωκυττάριου στρώματος (matrix) με ιδιότητες Th1 κυτοκίνης η οποία παράγεται από δενδριτικά κύτταρα και μακροφάγα (151). Προάγει την παραγωγή IFN-γ από τα Τ λεμφοκύτταρα και επάγει την Th2 ανταπόκριση (πολλαπλασιασμός Β κυττάρων, παραγωγή αντισωμάτων) μέσω αύξησης της έκφρασης CD40L από τα CD4 Τ λεμφοκύτταρα και αλληλεπίδρασης με τον υποδοχέα CD40 στα Β λεμφοκύτταρα (152). Τα επίπεδα της OPN έχουν βρεθεί αυξημένα σε ασθενείς με GD (153). Επιπρόσθετα, η προσθήκη ανασυνδυασμένης OPN ή πλάσματος από ασθενείς με GD σε καλλιέργειες CD4 λεμφοκυττάρων αύξησε την έκφραση CD40L (154), CCL20 και ΙL-17 από τα κύτταρα αυτά ενώ η προσθήκη αδρανοποιητικού αντι-OPN αντισώματος την ανέστειλε. Βαρύνουσας σημασίας είναι και η ανακάλυψη πως τα θυρεοκύτταρα φέρουν λειτουργικούς υποδοχείς ΙL-17 και δύνανται να παράγουν ΙL-6, CXCL10 και ICAM-1 ως απάντηση στην ΙL-17 που παράγεται από τα Τh17 λεμφοκύτταρα (155).
Πρόσφατες μελέτες έχουν αναδείξει το ρόλο των Τh22 λεμφοκυττάρων στην παθογένεια αρκετών αυτοάνοσων νοσημάτων (29,30). H IL-22, είτε παράγεται από τα Τh17 είτε από τα Th22 λεμφοκύτταρα φαίνεται να εμπλέκεται στην ανάπτυξη αυτοανοσίας (156, 157). Ειδικότερα, σε IL-22 knockout ποντίκια έχει παρατηρηθεί βελτίωση της αυτοανοσίας ενώ η αναστολή της IL-22 με τη χρήση αντισώματος είχε το ίδιο ευεργετικό αποτέλεσμα σε μοντέλα ποντικών (158). Όσον αφορά στον άνθρωπο, δύο μόνο μελέτες έχουν γίνει έως τώρα σε ασθενείς με GD, οι οποίες αναφέρουν αυξημένα ποσοστά περιφερικών Th22 και Th17 κυττάρων σε νεοδιεγνωσθέντες ασθενείς με GD καθώς και συσχέτιση των επιπέδων τους με τα διεγερτικά ΤSAb (133,159).
4.2.3. T fοllicular helper λεμφοκύτταρα
Η προσφάτως περιγραφείσα αυτή υποκατηγορία Τ λεμφοκυττάρων συμμετέχει στη ρύθμιση της αντιγονοειδικής ανοσίας των Β λεμφοκυττάρων και την παραγωγή αντισωμάτων. Τα Τfh λεμφοκύτταρα έχουν βρεθεί αυξημένα στη ΘΑ ενώ τα επίπεδά τους έχουν συσχετιστεί θετικά με τα επίπεδα των διεγερτικών TSAb, των anti-Tg και των anti-TPO αντισωμάτων. Σε μια υποκατηγορία ασθενών με GD τα επίπεδα Τfh υποχώρησαν μετά τη θεραπεία της GD. Όσον αφορά στην ΗΤ, έχει ανιχνευθεί παρουσία ενδοθυρεοειδικών Tfh κυττάρων, με αυξημένη έκφραση IL-21 και Bcl-6, σε δείγματα θυρεοειδικού ιστού από ασθενείς με ΗΤ (31,32).
4.2.4. Β λεμφοκύτταρα
Ιδιαίτερη σημασία έχει δοθεί στον πιθανό ρόλο της υποκατηγορίας των B ρυθμιστικών λεμφοκυττάρων (Β regulatory cells –Bregs) στην αιτιοπαθογένεια της ΘΑ. Τα κύτταρα αυτά εκφράζουν τα μόρια CD40, FCRL3, CD24hi, CD27 και SCGB3A2 και παράγουν IL-10. Τα Βregs έχουν ανοσοκατασταλτικό ρόλο, όπως και τα Tregs, και αναστέλλουν τον πολλαπλασιασμό των CD4 λεμφοκυττάρων και την παραγωγή ΤΝF-a, ΙFN-γ από αυτά. Τα επίπεδά τους έχουν βρεθεί μειωμένα σε ασθενείς με GD ενώ ανακάμπτουν μετά από θεραπεία της GD (160). Εκτός από τον αριθμό τους, μειωμένη βρέθηκε και η ικανότητά τους να καταστέλλουν τον πολλαπλασιασμό των CD4 λεμφοκυττάρων σε ασθενείς με GD (160).
Ένας άλλος παράγοντας που εκφράζουν τα Β λεμφοκύτταρα και έχει πρόσφατα εμπλακεί στην αιτιοπαθογένεια της ΘΑ είναι ο ΒΑFF (B lymphocyte activating factor), μια κυτοκίνη που ανήκει στην υπερ-οικογένεια του TNF και προάγει την παραγωγή αυτοαντισωμάτων με το να αυξάνει τον αριθμό και την ωρίμανση των Β λεμφοκυττάρων (161,162). Αυξημένα επίπεδα του BAFF έχουν ενοχοποιηθεί για την αυξημένη παραγωγή αντισωμάτων που παρατηρείται στο συστηματικό ερυθηματώδη λύκο, τη ρευματοειδή αρθρίτιδα και άλλα αυτοάνοσα νοσήματα. Όσον αφορά στη ΘΑ, αυξημένα επίπεδά του καθώς και θετική συσχέτισή τους με τα anti-Tg αντισώματα έχουν βρεθεί σε ασθενείς με GD (161).
Επιπρόσθετα, στην περίπτωση της GD, η έκφραση ΙCOSL παρατηρείται σε διηθούντα Β λεμφοκύτταρα αλλά και θυρεοκύτταρα, σε αντίθεση με την ΗΤ όπου παρατηρείται μόνο σε Β λεμφοκύτταρα (162). Η έκφρασή του ΙCOSL στα θυρεοκύτταρα επάγεται από τις ΙFN-γ, IL-6 και TNF-α και η αλληλεπίδρασή του με τον υποδοχέα ΙCOS συντελεί στην επικράτηση των θυρεοκυττάρων που χαρακτηρίζει την GD.
5. Ενδοθυρεοειδική είσοδος λεμφοκυττάρων και APC στη ΘΑ
Τόσο στην GD όσο και στην HT παρατηρείται είσοδος CD4 και CD8 λεμφοκυττάρων στο θυρεοειδικό παρέγχυμα με προεξάρχοντα τα πρώτα. Η μετανάστευση των λεμφοκυττάρων στο θυρεοειδή αδένα είναι αποτέλεσμα πολλαπλών μηνυμάτων προσέλκυσής τους (163). Έτσι στη ΘΑ παρατηρείται αύξηση της έκκρισης κυτοκινών όπως οι CCL3, CCL4, CXCL-10, CXCL-19 και παράγοντα RANTES (regulated upon activation, normal T-cell expressed and secreted) που προσελκύουν τα Τ λεμφοκύτταρα καθώς και αύξηση των αντίστοιχων υποδοχέων τους (CXCR3, RANTES receptor) στα ίδια τα λεμφοκύτταρα (163,164). Ακόμα ευρίσκεται αύξηση κυτοκινών με ρόλο στην οργάνωση των λεμφοειδών θυλακίων αλλά και άλλων χημειοτακτικών παραγόντων του ενδοθηλίου όπως των σελεκτινών (CLAA, E και P σελεκτίνες) και ιντεγκρινών (VCAM-1, ICAM-1) με τελικό αποτέλεσμα την αύξηση της προσκόλλησης των Τ λεμφοκυττάρων στις εξωκυττάριες πρωτεΐνες του ενδοθηλίου (165).
Μελέτες σε μοντέλα πειραματόζωων που αναπτύσσουν αυτοάνοση θυρεοειδίτιδα δείχνουν πως σε πρώτο στάδιο παρατηρείται μετανάστευση APC στο θυρεοειδή αδένα και διήθησή του (166). Τα φλεγμονώδη μηνύματα (κυτοκίνες) που οδηγούν στη συσσώρευση των APC μπορεί να οφείλονται σε νέκρωση ή βλάβη των θυρεοκυττάρων μετά από την επίδραση τοξινών, μικροβιακών παραγόντων ή περίσσειας ιωδίου. Επίσης μεταβολές του μεταβολισμού και του θυρεοειδικού μικροπεριβάλλοντος ίσως έχουν σημαντικό ρόλο. Ως επακόλουθο της φάσης αυτής τα εισβάλλοντα στο θυρεοειδή APC παρουσιάζουν θυρεοειδικά αυτοαντιγόνα στα Τ λεμφοκύτταρα.
6. Ενδοθυρεοειδική αλληλεπίδραση Τregs–APC–T λεμφοκυττάρων-Θυρεοκυττάρων στη ΘΑ
Στο στάδιο αυτό σημαντικός είναι ο ρόλος της περιφερικής ανοχής και της ρύθμισής της από συν-διεγερτικά μόρια, μια και αν αυτή παρακαμφθεί ανοίγει ο δρόμος για την ενεργοποίηση αυτοαντιδραστικών Τ λεμφοκυττάρων. H διατάραξη της ισορροπίας των προστατευτικών Tregs με τα ενδοθυρεοειδικά λεμφοκύτταρα και τα APC που συντελούν στη διαφοροποίηση των λεμφοκυττάρων προς την Th1 ή Τh2 κατεύθυνση, οδηγεί στην περαιτέρω επαγωγή της ΘΑ. Βασικό ρόλο στην ισορροπία αυτή φαίνεται πως έχει η IL-6, η οποία ωθεί την ανοσολογική απάντηση προς την Th17 ή Th1 κατεύθυνση παρά προς την επικράτηση της δράσης των Tregs (167). H δυσλειτουργία των Tregs, λόγω κυτοκινών που παράγονται από τα APC είτε τα ίδια τα Τ λεμφοκύτταρα, έχει ως αποτέλεσμα την αποδυνάμωση ή εξάλειψη της κατασταλτικής τους δράσης όσον αφορά την επιβίωση των αυτοαντιδραστικών Τ λεμφοκυττάρων. Τα Τ λεμφοκύτταρα τα οποία αλληλεπιδρούν με τα θυρεοειδικά αυτοαντιγόνα που τους παρουσιάζονται από τα APC μπορούν στη συνέχεια να ενεργοποιήσουν ελεύθερα την κυτταρική ή/και χυμική ανοσία. Τα θυρεοειδικά κύτταρα μπορούν και αυτά, λόγω της δράσης της παραγόμενης από τα Τ λεμφοκύτταρα IFN-γ, να εκφράσουν MHC μόρια και να δράσουν έτσι ως APC (168). Όσον αφορά την ικανότητα των θυρεοειδικών κυττάρων να εκφράζουν Β7.1 ή Β7.2 μόρια οι μελέτες εμφανίζουν αντικρουόμενα αποτελέσματα. Πάντως οι Battifora και συν. (140) ανακάλυψαν ισχυρή έκφραση Β7.1 στα θυρεοκύτταρα ασθενών με ΗΤ. Αν αυτό ισχύει, αλληλεπίδραση των θυρεοειδικών κυττάρων που φέρουν Β7.1 με Τ λεμφοκύτταρα που φέρουν CD28, θα μπορούσε να οδηγήσει στην επαγωγή της ενεργοποίησης Τ αυτοαντιδραστικών λεμφοκυττάρων. Ο ρόλος του sCTLA-4 δεν έχει ακόμα διευκρινισθεί μια και ορισμένες μελέτες βρίσκουν το μόριο αυτό αυξημένο σε ασθενείς με αυτοάνοσες θυρεοειδικές νόσους (18), ενώ άλλες μειωμένο (50).
Στο τελευταίο στάδιο, Τ και Β λεμφοκύτταρα διηθούν το θυρεοειδικό παρέγχυμα το οποίο μεταλλάσσεται σε “πεδίο μάχης” μεταξύ των θυρεοειδικών κυττάρων και των εισβαλλόντων λεμφοκυττάρων. Το είδος του κυττάρου που θα υπερισχύσει θα καθορίσει σε μεγάλο βαθμό το φαινότυπο της αυτοάνοσης θυρεοειδικής νόσου. Οι μηχανισμοί που εμπλέκονται στην καταστροφή των λεμφοκυττάρων/θυρεοκυττάρων στη ΘΑ περιλαμβάνουν την:
6.1. Κυτταρική ανοσία
- Κυτταροτοξικότητα εξαρτώμενη από αντισώματα (antibody dependent cell-mediated cytotoxicity). Τα κύτταρα του ανοσοποιητικού συστήματος (π.χ ΝΚ, μακροφάγα, ουδετερόφιλα) καταστρέφουν μέσω λύσης τα κύτταρα-στόχους (π.χ θυρεοκύτταρα) τα οποία έχουν επισημανθεί μέσω πρόσδεσης ειδικών αντισωμάτων στα αντιγόνα επιφανείας τους.
- Απόπτωση θυρεοκυττάρων ή λεμφοκυττάρων μέσω αλληλεπίδρασης των Fas/FasL (suicide/fratricide) και TRAIL/TRAIL-R που φέρουν στις επιφάνειές τους και των αντίστοιχων διαλυτών μορίων (sFas, sFasL, sTRAIL).
- Aμεση κυτταροτοξική δράση (homicide) των CD8 και CD4 κυττάρων (μέσω MHC-I και ΜΗC-II αντίστοιχα).
- Μονοπάτι κοκκίων perforins/granzyme των κυτταροτοξικών Τ και ΝΚ λεμφοκυττάρων.
- NK κύτταρα ενεργοποιημένα από κυτοκίνες.
6.2. Χυμική ανοσία
- Αντισώματα έναντι του TSH-R.
- Κυτταροτοξική δράση των anti-TPO αντισωμάτων μέσω συμπληρώματος.
Ωστόσο, μεταξύ των παραπάνω μηχανισμών, φαίνεται πως η απόπτωση μέσω αλληλεπίδρασης υποδοχέων και συνδετών θανάτου στην επιφάνεια των λεμφοκυττάρων και των θυρεοκυττάρων, έχει τον κυρίαρχο ρόλο.
7. Ο ρόλος της απόπτωσης στη ΘΑ
7.1. Αποπτωτικά μονοπάτια
Η απόπτωση ή προγραμματισμένος κυτταρικός θάνατος περιλαμβάνει την ενεργοποίηση ενός καταρράκτη πρωτεολυτικών ενζύμων (κασπάσες) με τελικό αποτέλεσμα την καταστροφή του κυττάρου. Η σύνδεση διαφορετικών υποδοχέων θανάτου με τους αντίστοιχους διαμεμβρανικούς συνδέτες τους αποτελεί ένα βασικό τρόπο έναρξης της μετάδοσης του αποπτωτικού σήματος στα κύτταρα που φέρουν τον υποδοχέα. Τα δύο πιο σημαντικά ζεύγη υποδοχέων-συνδετών που έχουν μελετηθεί σε σχέση με τη ΘΑ είναι το Fas/Fas ligand (FasL) και το DR4-DR5 (TRAILR-1, TRAILR-2) /TRAIL (TNF-related apoptosis inducing ligand) (2,97,169-171) (Εικόνα 2). Ο υποδοχέας Fas ανήκει στην οικογένεια των TNF υποδοχέων και σύνδεση του με τον FasL οδηγεί σε απόπτωση το κύτταρο που τον φέρει στην επιφάνεια του. Διαλυτές μορφές των Fas (sFas) και FasL (sFaL) έχουν επίσης αναγνωριστεί. To sFas συνδεόμενο με τον FasL, αναστέλλει την απόπτωση (172), ενώ το sFasL θεωρείται πως έπειτα από σύνδεσή του με τον Fas μπορεί να επάγει τους αποπτωτικούς μηχανισμούς (173) αν και οι μελέτες έχουν αντικρουόμενα αποτελέσματα. O συνδέτης TRAIL υπάρχει επίσης σε διαμεμβρανική και διαλυτή μορφή.
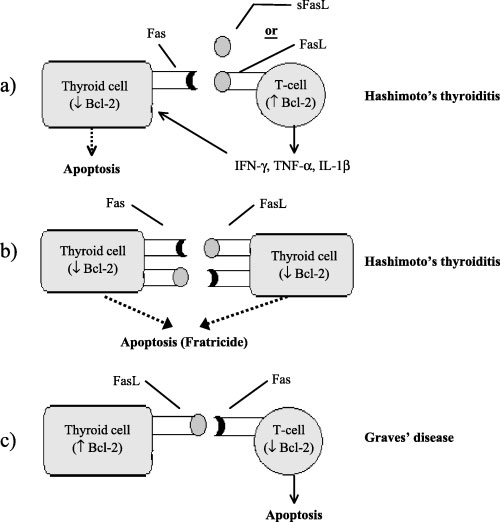
Εικόνα 2. Πιθανά μοντέλα για το ρόλο της Fas-επαγόμενης απόπτωσης στη ΘΑ. (a) Ενεργοποιημένα ενδοθυρεοειδικά Τ λεμφοκύτταρα παράγουν κυτοκίνες, οι οποίες επάγουν την ενεργοποίηση του αποπτωτικού μονοπατιού Fas στα θυρεοκύτταρα, οδηγώντας τα σε απόπτωση λόγω αλληλεπίδρασης με το FasL που εκφράζεται στην επιφάνεια των Τ λεμφοκυττάρων ή με το sFasL που προέρχεται από ενεργοποιημένα Τ λεμφοκύτταρα. (b) Ταυτόχρονη έκφραση Fas και FasL στην επιφάνεια των θυρεοκυττάρων μπορεί να οδηγήσει στην καταστροφή τους μέσω “αδελφοκτονίας”. (c) Σύνδεση του FasL, που εκφράζεται στην επιφάνεια των θυρεοκυττάρων, με τον υποδοχέα Fas που εκφράζεται σε ενδοθυρεοειδικά λεμφοκύτταρα μπορεί να οδηγήσει στον αποπτωτικό θάνατο των τελευταίων. Fountoulakis S & Tsatsoulis A, Clin Endocrinol (Oxf) 2004, with permission.
Η έκφραση των προαναφερθέντων διαμεμβρανικών μορίων από τα θυρεοειδικά κύτταρα εξακολουθεί να είναι πεδίο αντιπαραθέσεων. Σήμερα θεωρείται πως τα θυρεοειδικά κύτταρα εκφράζουν καθολικά και συνεχώς Fas και DR4, DR5, αλλά τα αποπτωτικά μονοπάτια που ξεκινούν από τους υποδοχείς αυτούς παραμένουν υπό φυσιολογικές συνθήκες ανενεργά (97, 174). Η αναστολή αυτή αίρεται από την επίδραση ορισμένων συνδυασμών προ-φλεγμονωδών κυτταροκινών (IFN-γ + IL-1β και IFN-γ + TNF-α) (139).
Τα δεδομένα για την έκφραση του FasL από τα θυρεοκύτταρα είναι λιγότερο σαφή, αλλά οι περισσότερες μελέτες συμφωνούν ότι τα θυρεοκύτταρα δεν εκφράζουν FasL, ίσως παρά μόνο σε παθολογικές καταστάσεις, “χρησιμοποιώντας” το για εξουδετερώσουν απειλητικά για αυτά κύτταρα. Υπάρχουν τέλος ενδείξεις πως η έκφραση αντιαποπτωτικών μορίων όπως το Bcl-2 μπορεί να χρησιμοποιηθεί είτε από τα θυρεοειδικά κύτταρα είτε από τα λεμφοκύτταρα ως μέσο επιβίωσης (174,175).
7.2. Αλληλεπίδραση αυτοαντιδραστικών λεμφοκυττάρων-θυρεοκυττάρων στη ΘΑ- Απόπτωση και κλινική έκφραση ΘΑ
7.2.1. Θυρεοειδίτιδα Hashimoto
Η απόπτωση θεωρείται ως ο βασικός μηχανισμός καταστροφής των θυρεοκυττάρων που παρατηρείται στην ΗΤ (2). Δύο είναι οι κύριες θεωρίες που έχουν διατυπωθεί. Η πρώτη, που φαίνεται να κερδίζει έδαφος σύμφωνα με τα τελευταία δεδομένα, υποστηρίζει πως ο αποπτωτικός θάνατος των θυρεοκυττάρων επάγεται όταν ενδοθυρεοειδικά λεμφοκύτταρα που εκφράζουν FasL και ταυτόχρονα Bcl-2 έρχονται σε επαφή με τον Fas υποδοχέα των θυρεοκυττάρων (2). Τα λεμφοκύτταρα μετά την ενεργοποίησή τους από APC που φέρουν Β7.1 συνδιεγερτικά μόρια ή και από τα ίδια τα θυρεοκύτταρα που φέρουν Β7.1 (11), εκκρίνουν τοπικά Τh1 κυτταροκίνες. Οι τελευταίες άρουν την αναστολή του Fas/FasL μονοπατιού, η οποία υπάρχει φυσιολογικά στα θυρεοκύτταρα, καθιστώντας τα ευάλωτα σε απόπτωση.
Από την άλλη πλευρά οι Giordano et al., (175) υποστηρίζουν έναν μηχανισμό “αδελφοκτονίας” (fratricide) των θυρεοκυττάρων λόγω ταυτόχρονης έκφρασης Fas και FasL στην επιφάνεια τους. Η θεωρία αυτή δέχεται πως τα θυρεοκύτταρα μπορούν να εκφράζουν FasL το οποίο είναι ενεργό και λειτουργικό, κάτι με το οποίο πάντως δε συμφωνούν οι περισσότερες μελέτες.
Άλλα μόρια που εμπλέκονται στην παθογένεια της ΗΤ είναι τα διαλυτά sFas και sFasL. Το προερχόμενο από τα λεμφοκύτταρα sFasL μπορεί να επάγει την απόπτωση σε κύτταρα που φέρουν Fas (175). Επίσης τα επίπεδα του sFas έχουν βρεθεί μειωμένα στη ΗΤ, οδηγώντας στην υπόθεση πως το ακέραιο διαμεμβρανικό Fas, από το οποίο προέρχονται, είναι αυξημένο και επάγει την απόπτωση των θυρεοκυττάρων που το φέρουν. Ακόμα υπάρχουν ενδείξεις πως η έκφραση του προστατευτικού μορίου Bcl-2, είναι μειωμένο στα θυρεοκύτταρα ασθενών με ΗΤ (175,176) (Εικόνα 3).
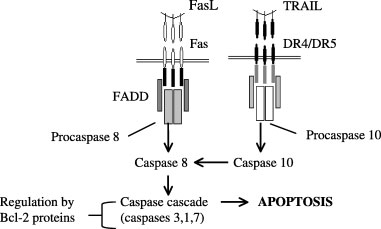
Εικόνα 3. Συστατικά στοιχεία των Fas και DR4/DR5 αποπτωτικών μονοπατιών. Μετά από τη σύνδεση με το FasL, ο υποδοχέας Fas τριμερίζεται και επιστρατεύει, μέσω του πεδίου (domain) θανάτου, την πρωτεινη FADD (Fas-associated protein with death domain). Αυτή με τη σειρά της επιστρατεύει και αλληλεπιδρά με την προκασπάση 8. Ο σχηματισμός του συμπλέγματος Fas-FADD-προκασπάσης 8 ευοδώνει την ενεργοποίηση της προκασπάσης 8. Η κασπάση 8 ενεργοποιεί την κασπάση 3 καθώς και άλλες κασπάσες. Ο καταρράκτης διαδοχικής ενεργοποίησης των κασπασών οδηγεί τελικά στην απόπτωση του κυττάρου. Το TRAIL με τη σειρά του, προσδένεται στους επιφανειακούς υποδοχείς DR4 και DR5, οι οποίοι επιστρατεύουν την προκασπάση 10 με επακόλουθο την ενεργοποίησή της. Στη συνέχεια η κασπάση 10 ενεργοποιεί την κασπάση 8, δίνοντας έναυσμα για τον αποπτωτικό καταρράκτη των κασπασών. Μέλη της υπεροικογένειας του Bcl-2 μπορούν να δράσουν ως ρυθμιστές της απόπτωσης. Fountoulakis S & Tsatsoulis A, Clin Endocrinol (Oxf) 2004, with permission.
Σημαντικό ρόλο ακόμα διεκδικεί η ευαισθητοποίηση των θυρεοκυττάρων σε TRAIL/DR4-DR5 απόπτωση από τις κυτταροκίνες των Τ λεμφοκυττάρων (2). Τέλος η απώλεια ή μείωση της επικοινωνίας των γειτονικών θυρεοκυττάρων μέσω των χασματικών ενώσεων (gap junctions), που έχει παρατηρηθεί σε πειραματικά μοντέλα θυρεοειδικής αυτοανοσίας ίσως οδηγεί σε έναν τύπο αποπτωτικού θανάτου με την ελληνική ονομασία anoikis (άνευ οίκου) (177).
7.2.2. Nόσος του Graves
Στη GD φαίνεται πως επικρατούν μηχανισμοί επιβίωσης των θυρεοκυττάρων, ίσως λόγω της επικράτησης των Th2 κυττοκινών, προερχόμενων από Β7.2 ενεργοποίηση των λεμφοκυττάρων (2,178). Έτσι υπερισχύει η χυμική ανοσία και η παραγωγή αυτοαντισωμάτων που διεγείρουν τον υποδοχέα της TSH, οδηγούν σε υπερπλασία των θυρεοκυττάρων και ίσως έχουν και αντιαποπτωτικό ρόλο. Επίσης παρατηρείται αύξηση του Bcl-2 στα θυρεοκύτταρα, ενώ η έκφραση μορίων όπως το FasL και το TRAIL μπορεί να επάγεται στην επιφάνεια τους (2,170,178), αποτελώντας ένα ισχυρό όπλο στη μάχη ενάντια των λεμφοκυττάρων, τα οποία εκφράζουν Fas και μειωμένα επίπεδα FasL και Bcl-2.
Περνώντας στην ατροφική θυρεοειδίτιδα αξίζει να τονιστεί πως η υποκατηγορία ασθενών που φέρουν ανταγωνιστικά προς τον υποδοχέα της TSH αντισώματα μπορεί να θεωρηθεί ως το αντίθετο άκρο της GD. Kαι εδώ έχουμε επικράτηση της χυμικής ανοσίας, ενώ ο παθογενετικός ρόλος των ανταγωνιστικών anti-TSH αντισωμάτων ενισχύεται από την ικανότητά τους να προκαλούν παροδικό νεογνικό υποθυρεοειδισμό (179).
Τέλος οι υπόλοιπες μορφές της χρόνιας αυτοάνοσης θυρεοειδίτιδας (σποραδική θυρεοειδίτιδα και θυρεοειδίτιδα της λοχείας) ομοιάζουν με τη ΗΤ, μόνο που η λεμφοκυτταρική διήθηση είναι ηπιότερη. Η αρχική φάση χαρακτηρίζεται από καταστροφή των θυρεοκυττάρων και ήπια θυρεοτοξίκωση αλλά είναι παροδική και ακολουθείται κάποιες φορές από την υποθυρεοειδική φάση ανάρρωσης που και αυτή συνήθως είναι παροδική με τον ασθενή να επιστρέφει σταδιακά σε ευθυρεοειδική κατάσταση.
8. Επιγραμματικά
Η έναρξη και η εξέλιξη της ΘΑ είναι αποτέλεσμα επίδρασης περιβαλλοντικών παραγόντων σε γενετικά προδιατεθειμένα άτομα.
- Η ύπαρξη και η αλληλεπίδραση πολυμορφισμών προδιαθεσικών γονιδίων (όπως τα HLA-DR3, CTLA-4, FCRL3, CD40, PTPN22, ΙL2Ra/ CD25, TSHR, Tg, SCGB3A2 κ.α) θέτουν τη βάση ανάπτυξης της ΘΑ. Στο αρχικό αυτό υπόβαθρο ίσως επιδρούν και άλλοι παράγοντες όπως τα μειωμένα επίπεδα miRNA, τα αυξημένα DAMPs και ο μικροχιμαιρισμός. O ρόλος των miRNA έχει να κάνει με την επίδρασή τους σε γονίδια-στόχους (target genes) όπως γονίδια χυμοκινών και άλλα γονίδια σηματοδοτικών μονοπατιών κυτοκινών. Από την άλλη πλευρά τα εμβρυικά μικροχιμαιρικά Τ, ΝΚ, και μονοκύτταρα/μακροφάγα μπορεί να εμπλέκονται στην έναρξη ΘΑ μέσω των κυτοκινών που παράγουν ή να αναγνωρίζονται τα ίδια ως ‘μερικώς’εξωγενή. Επιπρόσθετα, υπό διερεύνηση είναι ο ρόλος των DAMPs που παράγονται από θνήσκωντα κύτταρα, εισέρχονται στα θυρεοκύτταρα και παρουσιάζονται από MHC-II στην επιφάνεια των θυρεοκυττάρων σε CD4 λεμφοκύτταρα πυροδοτώντας έτσι την έναρξη ανοσολογικής απάντησης.
- Στο υπόβαθρο αυτό επιδρούν περιβαλλοντικοί παράγοντες όπως το κάπνισμα ή το ιώδιο που αυξάνει την αντιγονικότητα της Tg και πιθανόν προκαλεί την απελευθέρωση θυρεοειδικών αυτοαντιγόνων. Επίσης ρόλο ίσως έχουν και μικροβιακοί οργανισμοί, που μέσω του μηχανισμού της μοριακής μίμησης ενεργοποιούν το ανοσολογικό οπλοστάσιο. Ακόμα το stress μπορεί μέσω διαφορετικών μηχανισμών (κυτοκίνες, μείωση DHEAS) να ευοδώσει την Th2/Th17 ανταπόκριση και ακολούθως τη χυμική ανοσία και παραγωγή αυτοαντιδραστικών αντισωμάτων ευνοώντας την εμφάνιση GD. Επίσης τα μειωμένα επίπεδα σεληνίου ίσως συνεισφέρουν στην ανάπτυξη ΘΑ, ιδιαίτερα μετά την ανακάλυψη πως η χορήγηση σεληνίου αυξάνει τον αριθμό Tregs σε μοντέλα αυτοάνοσης θυρεοειδίτιδας και μειώνει την απελευθέρωση Th1 κυτοκινών από τα περιφερικά λεμφοκύτταρα ευθυρεοειδικών ασθενών με ΗΤ.
- Τόσο στη GD όσο και στην HT παρατηρείται είσοδος CD4, CD8 λεμφοκυττάρων και APC στο θυρεοειδικό παρέγχυμα με προεξάρχοντα τα πρώτα. Η μετανάστευση των λεμφοκυττάρων στο θυρεοειδή αδένα είναι αποτέλεσμα πολλαπλών μηνυμάτων προσέλκυσης. Τα Τ λεμφοκύτταρα και τα APC εισέρχονται στο θυρεοειδή ως αποτέλεσμα αύξησης της έκκρισης κυτοκινών και άλλων χημειοτακτικών παραγόντων του ενδοθηλίου. Ως επακόλουθο, τα εισβάλλοντα στο θυρεοειδή APC παρουσιάζουν θυρεοειδικά αυτοαντιγόνα στα Τ λεμφοκύτταρα.
- Tregs και Bregs
– Εξαιρετικά σημαντικό ρόλο στη διαφυγή από την περιφερική ανοσολογική ανοχή και την προαγωγή ή αποτροπή των δύο επόμενων σταδίων έχει η δράση των Tregs που διηθούν το θυρεοειδή και παράγουν ΙL-10 και TGF-b. Ρόλος των Tregs είναι ο έλεγχος και η καταστολή των αυτοαντιδραστικών λεμφοκυττάρων (Τ effectors) που μπορεί να εμφανιστούν. Ωστόσο στη ΘΑ τα κυκλοφορούντα Τregs είναι δυσλειτουργικά και λιγότερο ικανά να αναστείλουν τον πολλαπλασιασμό των T αυτοαντιδραστικών λεμφοκυττάρων (Τ effectors) με τελικό αποτέλεσμα την απώλεια ανοχής έναντι των θυρεοειδικών αντιγόνων και εμφάνιση αντισωμάτων (π.χ anti-TSHR). Η δυσλειτουργία ή η πιθανή μείωση του αριθμού των Tregs στη ΘΑ ίσως οφείλεται στην παραγωγή IFN-a από τα APC (δενδριτικά κύτταρα) στη GD. Στο στάδιο αυτό, το αποτέλεσμα της “μάχης” των Tregs με τα T effectors (Τh1, Th2 ή Th17) θα καθορίσει τη διήθηση ή όχι του θυρεοειδούς από άλλοτε μεγάλο και άλλοτε μικρότερο αριθμό Β και Τ λεμφοκυττάρων.
– Τελευταία έχει αναδειχθεί και ο ρόλος των Bregs στη ΘΑ λόγω της ανοσοκατασταλτικής τους δράσης στον πολλαπλασιασμό των CD4 λεμφοκυττάρων και την παραγωγή ΤΝF-a, ΙFN-γ από αυτά. Στη GD, τα επίπεδά τους και η ικανότητά τους να καταστέλλουν τον πολλαπλασιασμό των CD4 λεμφοκυττάρων είναι μειωμένα. - Το επόμενο στάδιο στην αιτιοπαθογένεια της ΘΑ έχει να κάνει με το αποτέλεσμα της επίδρασης των γενετικών και περιβαλλοντικών παραγόντων στην αντιγονοπαρουσίαση ανοσογονικών επιτόπων των θυρεοειδικών αυτοαντιγόνων (π.χ Tg) από τα APC στα ενδοθυρεοειδικά CD4 βοηθητικά Τ λεμφοκύτταρα. Μελέτες σε μοντέλα ποντικών και ασθενείς με ΘΑ έχουν δείξει πως τα CD4 Τ λεμφοκύτταρα, ανάλογα με το είδος του συνδιεγερτικού μορίου (Β7.1, Β7.2, CTLA-4) και τις κυτοκίνες που δέχονται από τα APC (ή τα θυρεοκύτταρα που μπορούν να δρουν ως APC), μπορούν να διαφοροποιηθούν σε Th1, Τh2 ή Th17. Σημαντική είναι και η ανεύρεση CD8 κυτταροτοξικών λεμφοκυττάρων αντιγονοειδικών για Tg/TPO στο περιφερικό αίμα ασθενών σε αρχικό στάδιο ΗΤ.
- Στη συνέχεια τα CD4 λεμφοκύτταρα (Τh1 ή Th2 ή Th17) που διέφυγαν της ανοσολογικής ανοχής (Τ effectors) διηθούν το θυρεοειδή αδένα με προεξάρχοντα τα Τh1/Th17 στην ΗΤ και τα Τh2 στην GD. Nεότερες μελέτες αναδεικνύουν τον πιθανό ρόλο και των Th1 στη GD.
– Στην ΗΤ, τα APC εκφράζουν κυρίως B7.1 με αποτέλεσμα να προσφέρουν στα- διηθούντα το θυρεοειδή- λεμφοκύτταρα συνδιεγερτικό σήμα για την παραγωγή κυρίως Th1 κυτοκινών. Αντίθετα στη GD παρατηρείται κυρίως Th2 ανταπόκριση ως αποτέλεσμα της B7.2 συνδιέγερσης των λεμφοκυττάρων και παραγωγή κυρίως Th2 κυτοκινών. Φαίνεται πως στη GD, οι Th2 κυτοκίνες μπορούν να μεταβάλλουν και την έκφραση του MHC-II στα θυρεοκύτταρα ενισχύοντας την παρουσίαση αυτοαντιγόνων (TSH-R) από τα θυρεοκύτταρα στα λεμφοκύτταρα. Αξίζει να παρατηρηθεί πως όταν τα θυρεοκύτταρα διεγερθούν από IFN-γ (Th1) παράγουν την κυτοκίνη CXCL10 (Interferon gamma-induced protein 10 ή ΙP10) που στρατολογεί Th1 κύτταρα τα οποία παράγουν CXCR3 (υποδοχέα της CXCL10) και IFN-γ οδηγώντας έτσι σε φαύλο κύκλο.
– Tα αυξημένα επίπεδα των Th22 και Τh17 κυττάρων έχουν εμπλακεί στην αιτιοπαθογένεια της HT. Η ΤSLP που παράγεται από ΑPC μπορεί να ενεργοποιήσει τα CD4 λεμφοκύτταρα προς την Th2 και την Τh17 κατεύθυνση. Στην GD τα αυξημένα επίπεδα CCL20 και OPN, που παράγεται από τα APC, ευοδώνουν τη μετάδοση σήματος της IL-17 και επάγουν την Th2 ανταπόκριση.
– Η παραγωγή αντισωμάτων από τα Β λεμφοκύτταρα αποτελεί ένα ακόμη βασικό στάδιο της αιτιοπαθογένειας της ΘΑ, κυρίως όσον αφορά στη GD. H αλληλεπίδραση του CD40L που εκφράζεται στα Τh2 λεμφοκύτταρα, λόγω της αυξημένης ΟPN με τον CD40 των Β λεμφοκυττάρων, οδηγεί σε ενεργοποίηση των τελευταίων και παραγωγή αντισωμάτων. Επίσης τα αυξημένα επίπεδά του παράγοντα ΒΑFF, που εκφράζουν τα Β λεμφοκύτταρα, προάγουν την παραγωγή αυτοαντισωμάτων μέσω αύξησης του αριθμού και της ωρίμανσης των Β λεμφοκύτταρων. Επιπρόσθετα, τα αυξημένα επίπεδα των Tfh λεμφοκυττάρων συμμετέχουν στη ρύθμιση της αντιγονοειδικής ανοσίας των Β λεμφοκυττάρων και την παραγωγή αντισωμάτων. - Tα Τ αυτοαντιδραστικά (Τ effector) λεμφοκύτταρα (Τh1, Th2 ή Th17) που θα διαφύγουν τελικά από τη ρυθμιστική δράση των Τregs και Βregs αλληλεπιδρούν με τα θυρεοειδικά κύτταρα σε μια δεύτερη “μάχη για επιβίωση”. H έκβαση της μάχης αυτής θα καθορίσει την φαινοτυπική έκφραση της ΘΑ. Σημαντικός είναι ο ρόλος των διαφορών στο θυρεοειδικό μικροπεριβάλλον, που περιλαμβάνει συν-διεγερτικά μόρια (Β7.1 ή Β7.2), κυτταροκίνες (Th1, Th2 ή Τh17), υποδοχείς και συνδέτες θανάτου (Fas, FasL, DR-4/DR-5, TRAIL), αντιαποπτωτικά μόρια (Bcl-2) και διαλυτά μόρια (sFas, sFasL). Στη GD φαίνεται πως επικρατούν μηχανισμοί επιβίωσης των θυρεοκυττάρων, ίσως λόγω της επικράτησης των Th2 κυτοκινών. Έτσι παρατηρείται αύξηση του Bcl-2, ενώ η έκφραση μορίων όπως το FasL και το TRAIL μπορεί να επάγεται στην επιφάνειά τους με τελικό αποτέλεσμα την επιβίωσή τους και την υπερίσχυση της χυμικής ανοσία και της παραγωγής αυτοαντισωμάτων που διεγείρουν τον υποδοχέα της TSH και οδηγούν σε υπερπλασία των θυρεοκυττάρων.
Στην ΗΤ ο αποπτωτικός θάνατος των θυρεοκυττάρων επάγεται όταν ενδοθυρεοειδικά λεμφοκύτταρα που εκκρίνουν τοπικά Τh1 κυτοκίνες, εκφράζουν FasL και Bcl-2 και ίσως εκκρίνουν sFasL, έρχονται σε επαφή με τον Fas υποδοχέα των θυρεοκυττάρων. Τα τελευταία εκφράζουν μειωμένο Bcl-2 και αυξημένο Fas. Οι Τh1 κυτοκίνες ίσως άρουν την αναστολή του Fas/FasL μονοπατιού, η οποία υπάρχει φυσιολογικά στα θυρεοκύτταρα, καθιστώντας τα έτσι ευάλωτα σε απόπτωση. Αξίζει τέλος να σημειωθεί και η υπόθεση των Giordano et al, (175) περί υπάρξεως ενός μηχανισμό “αδελφοκτονίας” των θυρεοκυττάρων στην ΗΤ λόγω ταυτόχρονης έκφρασης Fas και FasL στην επιφάνεια τους.
Ολοκληρώνοντας, γίνεται εμφανές πως η αιτιοπαθογένεια της ΘΑ είναι πολυπαραγοντική με πολλαπλούς συντελεστές να αλληλεπιδρούν και να συνεισφέρουν σε ποικίλο βαθμό στα διαφορετικά στάδια ανάπτυξης της νόσου. Η ανάγκη ποσοτικοποίησης της συνεισφοράς των προαναφερθέντων μηχανισμών στην έναρξη και εξέλιξη των σταδίων της ΘΑ ανακύπτει ως ιδιαίτερα σημαντική. Ακόμα ο ακριβής ρόλος των νέων μορίων που φαίνονται να εμπλέκονται στην παθογένεια της ΘΑ χρειάζεται αποσαφήνιση. Τέλος το μεγάλο ερώτημα γιατί και πότε εμφανίζονται αυτοαντιγόνα παραμένει ακόμα αναπάντητο.
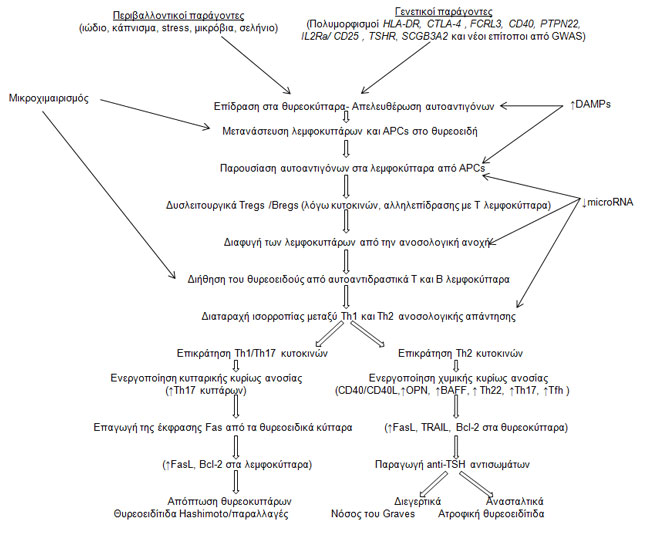
Εικόνα 4. Σύνοψη της αιτιοπαθογένειςα της θυρεοειδικής αυτοανοσίας.
Βιβλιογραφία
1. McGrogan A, Seaman H, Wright J, de Vries C. The incidence of autoimmune thyroid disease: a systematic review of the literature. Clin Endocrinol (Oxf) 2008; 69:687.
2. Fountoulakis S, Tsatsoulis A. On the pathogenesis of AITD: a unifying hypothesis. Clin Endocrinol (Oxf) 2004; 60:397.
3. Kemp E, Sandhu H, Watson P, Weetman A. Low frequency of pendrin autoantibodies detected using a radioligand binding assay in patients with autoimmune thyroid disease. J Clin Endocrinol Metab 2013; 98:E309.
4. Rotondi M, Chiovato L. The chemokine system as a therapeutic target in autoimmune thyroid diseases: a focus on the interferon-γ inducible chemokines and their receptor. Curr Pharm Des 2010; 17:3202.
5. Ganesh BB, Bhattacharya P, Gopisetty A, Prabhakar BS. Role of cytokines in the pathogenesis and suppression of thyroid autoimmunity. J Interferon Cytokine Res 2011; 31: 721.
6. Tunbridge WM, Evered DC, Hall R, Appleton D, Brewis M, Clark F et al. The spectrum of thyroid disease in a community: the Whickham survey. Clin Endocrinol (Oxf) 1977; 7:481.
7. Hollowell G et al. Serum TSH, T4 and thyroid antibodies in the United States population (1984 to 1994): National Health and nutritio Examination Survey (NHANES III). J Clin Endocrinol Metab 2002; 87:489.
8. Katz SM, Vickery AL Jr. The fibrous variant of Hashimoto’s thyroiditis. Hum Pathol 1974; 5: 161.
9. Orgiazzi J. Anti-TSH receptor antibodies in clinical practice. Endocrinol and Metab Clin North Am 2000; 29: 339.
10. Ahmed R, Al-Shaikh S, Akhtar M. Hashimoto thyroiditis: a century later. Adv Anat Pathol 2012 ;19:181.
11. Kennichi Kakudo et al. IgG4-related disease of the thyroid glands. Endocrine Journal 2012; 59:273.
12. Umehara H et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol 2012; 22:1.
13. Canaris GJ et al. The Colorado thyroid disease prevalence study. Arch Intern Med 2000; 160:526.
14. Kek PC et al. Subclinical thyroid disease. Singapore Med J 2003; 44:595.
15. Bluher M et al. Cytokine gene expression in autoimmune thyroiditis in BioBreeding/Worcester rats. Thyroid 1999; 9: 1049.
16. McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity 2008 ; 28: 445.
17. Salmaso C et al. Co-stimulatory molecules and autoimmune thyroid diseases. Autoimmunity 2002; 35: 159.
18. Oaks M et al. A native soluble form of CTLA-4. Cell Immunol 2000; 201: 144.
19. Guo J, Stolina M, Bready J et al. Stimulatory effects of B7-related Protein-1 on cellular and humoral immune responses in mice. J Immunol 2001; 166: 5578.
20. Brown J, Dorfman D, Ma F et al. Blockade of programmed deaTh1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol 2003; 170: 1257.
21. Jiang H, Chess L. Mechanisms of Disease Regulation of Immune Responses by T Cells. NEJM 2006; 354:1166.
22. Shevach EM. From Vanilla to 28 Flavors: Multiple Varieties of T Regulatory Cells. Immunity 2006; 25:195.
23. Takahashi T et al. Dominant immunologic self-tolerance by CD25+CD4+ regulatory T cells constitutively expressing CTLA-4. J Exp Med 2000; 192:303.
24. Kolls J, Linden N. Interleukin-17 family members and inflammation. Immunity 2004; 21: 467.
25. Yen D, Cheung J, Scheerens H et al. IL -23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 2006; 116: 1310.
26. Bettelli E, Carrier Y, Gao W et al. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory Tcells. Nature 2006; 441: 235.
27. Eyerich S, Eyerich K, Cavani A et al. IL-17 and IL-22: siblings, not twins. Trends Immunol 2010; 31:354.
28. Sanjabi S, Zenewicz LA, Kamanaka M et al. Anti-inflammatory and proinflammatory roles of TGF-b, IL-10, and IL-22 in immunity and autoimmunity. Curr Opin Pharmacol 2009; 9: 447.
29. Wolk K, Witte E, Witte K et al. Biology of interleukin-22. Semin Immunopathol 2010; 32:17.
30. Eyerich S, Eyerich K, Pennino D et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 2009; 119:3573.
31. Kim C, Lim H, Kim J et al. Unique gene expression program of human germinal center T helper cells. Blood 2004; 104: 1952.
32. Breitfeld D, Ohl L, Kremmer E et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med 2000; 192: 1545.
33. Schwartz R. T cell anergy. Ann Rev Immunol 2003; 21: 305.
34. Anderson M. Update in endocrine autoimmunity. J Clin Endocrinol Metab 2008; 93: 3663.
35. Cetani F, Barbesino G, Borsari S et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab 2001; 86: 4747.
36. Diamond M, Joffe B. Monozygotic twins with chronic lymphocytic thyroiditis. (Hashimoto’s disease). JAMA 1966; 198: 182.
37. Brix T, Kyvik K, Christensen K. Evidence for a major role of heredity in Graves’ disease: a population-based study of two Danish twin cohorts. J Clin Endocrinol Metab 2001; 86: 930.
38. Brand O, Gough S. Immunogenetic Mechanisms Leading to Thyroid Autoimmunity: Recent Advances in Identifying Susceptibility Genes and Regions. Current Genomics 2011; 12: 526.
39. Tomer Y. Genetic Susceptibility to Autoimmune Thyroid Disease: Past, Present, and Future. Thyroid 2010; 20: 715.
40. International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449:851.
41. International HapMap Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467:52.
42. Wellcome Trust Case Control Consortium. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Gen 2007; 39: 1329.
43. Zamani M, Spaepen M, Bex M. Primary role of the HLA class II DRB1*0301 allele in Graves disease. Am J Med Genet 2000; 95:432.
44. Ban Y, Davies T, Greenberg D et al. Arginine at position 74 of the HLA-DRb1 chain is associated with Graves’ disease. Genes Immun 2004; 5:203.
45. Menconi F, Monti M, Greenberg D et al. Molecular amino acid signatures in the MHC class II peptide-binding pocket predispose to autoimmune thyroiditis in humans and in mice. Proc Natl Acad Sci U SA 2008; 105:14034.
46. Simmonds M, Howson J, Heward J et al. A novel and major association of HLA-C in Graves’ disease that eclipses the classical HLA-DRB1 effect. Hum. Mol. Genet. 2007; 16:2149.
47. Du L, Yang J, Huang J et al. The associations between the polymorphisms in the CTLA-4 gene and the risk of Graves’ disease in the Chinese population. BMC Med Genet 2013; 19:14.
48. Kotsa K, Watson P, Weetman AP. A CTLA-4 gene polymorphism is associated with both Graves’ disease and autoimmune hypothyroidism. Clin Endocrinol 1997; 46:551.
49. Kouki T, Gardine CA, Yanagawa T et al. Relation of three polymorphisms of the CTLA-4 gene in patients with Graves’ disease. J Endocrinol Invest 2002; 25:208.
50. Ueda H, Howson J, Esposito L et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003; 423:506.
51. Ban Y, Davies T, Greenberg D et al. Analysis of the CTLA-4, CD28 and inducible co-stimulator (ICOS) genes in autoimmune thyroid disease. Genes Immun 2003; 4:586.
52. Kouki T, Sawai Y, Gardine C et al. CTLA-4 gene polymorphism at position 49 in exon 1 reduces the inhibitory function of CTLA-4 and contributes to the pathogenesis of Graves’ disease. J Immunol 2000; 165:6606.
53. Wang X, Kakoulidou M, Giscombe R et al. Abnormal expression of CTLA-4 by T cells from patients with myasthenia gravis: effect of an AT-rich gene sequence. J Neuroimmunol 2002; 130:224.
54. Zhao SX, Liu W, Zhan M et al. China Consortium for the Genetics of Autoimmune Thyroid Disease A refined study of FCRL genes from a genome-wide association study for Graves’ disease. PLoS One 2013; 8:e57758.
55. Yang J, Qin Q, Yan N et al. CD40 C/T(-1) and CTLA-4 A/G(49) SNPs are associated with autoimmune thyroid diseases in the Chinese population. Endocrine 2012; 41:111.
56. Li M, Sun H, Liu S et al. CD40 C/T-1 polymorphism plays different roles in Graves’disease and Hashimoto’s thyroiditis: a meta-analysis. Endocrine Journal 2012; 59: 1041.
57. Pearce SH, Vaidya B, Imrie H et al. Further evidence for a susceptibility locus on chromosome 20q13.11 in families with dominant transmission of Graves disease. Am J Hum Genet 1999; 65:1462.
58. Ban Y, Tozaki T, Taniyama M et al. Association of a C/T single-nucleotide polymorphism in the 50 untranslated region of the CD40 gene with Graves’ disease in Japanese. Thyroid 2006; 16:443.
59. Jacobson E, Concepcion E, Oashi T et al. A Graves’ disease-associated Kozak sequence single-nucleotide polymorphism enhances the efficiency of CD40 gene translation: a case for translational pathophysiology. Endocrinology 2005; 146:2684.
60. Luo L, Cai B, Liu F et al. Association of Protein Tyrosine Phosphatase Nonreceptor 22 (PTPN22) C1858T gene polymorphism with susceptibility to autoimmune thyroid diseases: a meta-analysis. Endocr J 2012; 59:439.
61. Vang T, Congia M, Macis M et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet 2005; 37:1317.
62. Criswell L, Pfeiffer K, Lum R. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet 2005; 76:561.
63. Brand O, Lowe C, Heward J et al. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves’ disease using a multilocus test and tag SNPs. Clin Endocrinol (Oxf) 2007; 66:508.
64. Fountoulakis S, Vartholomatos G, Kolaitis N. HLA-DR expressing peripheral T regulatory cells in newly diagnosed patients with different forms of autoimmune thyroid disease. Thyroid 2008; 18:1195.
65. Liu L, Wu HQ, Wang Q et al. Association between thyroid stimulating hormone receptor gene intron polymorphisms and autoimmune thyroid disease in a Chinese Han population Endocr J 2012; 59:717.
66. Dechairo B, Zabaneh D, Collins J et al. Association of the TSHR gene with Graves’ disease: the first disease specific locus. Eur J Hum Genet 2005; 13:1223.
67. Yin X, Latif R, Bahn R et al. Influence of the TSH receptor gene on susceptibility to Graves’ disease and Graves’ ophthalmopathy. Thyroid 2008; 18:1201.
68. Brand O, Barrett J, Simmonds M et al. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves’ disease. Hum Mol Genet 2009; 18: 1704.
69. Sakai K, Shirasawa S, Ishikawa N et al. Identification of susceptibility loci for autoimmune thyroid disease to 5q31-q33 and Hashimoto’s thyroiditis to 8q23-q24 by multipoint affected sib-pair linkage analysis in Japanese. Hum Mol Genet 2001; 10: 1379.
70. Song H, Liang J, Shi J et al. Functional SNPs in the SCGB3A2 promoter are associated with susceptibility to Graves’ disease. Hum Mol Genet 2009; 18:1156.
71. Chistiakov D, Voronova N, Turakulov R. The -112G > A polymorphism of the secretoglobin 3A2 (SCGB3A2) gene encoding uteroglobin-related protein 1 (UGRP1) increases risk for the development of Graves’ disease in subsets of patients with elevated levels of immunoglobulin E. J. Appl. Genet 2011; 52:201.
72. Chu X, Dong C, Lei R et al. Polymorphisms in the interleukin 3 gene show strong association with susceptibility to Graves’ disease in Chinese population. Genes Immun 2009; 10: 260.
73. Ban Y, Tozaki T, Taniyama M et al. Multiple SNPs in intron 41 of thyroglobulin gene are associated with autoimmune thyroid disease in the Japanese population. PLoS One 2012; 7:e37501.
74. Collins J, Heward J, Carr-Smith J et al. Association of a rare thyroglobulin gene microsatellite variant with autoimmune thyroid disease. J Clin Endocrinol Metab 2003; 88:5039.
75. Ban Y, Greenberg DA, Concepcion E et al. Amino acid substitutions in the thyroglobulin gene are associated with susceptibility to human and murine autoimmune thyroid disease. Proc Natl Acad Sci USA 2003; 100:15119.
76. Cooper J, Simmonds M, Walker N et al. Wellcome Trust Case Control Consortium. Seven newly identified loci for autoimmune thyroid disease Hum Mol Genet 2012; 21: 5202.
77. Chu X, Pan C, Zhao S et al. A genome-wide association study identifies two new risk loci for Graves’ disease. Nat Genet 2011; 43: 897.
78. Zhao S, Xue L Liu W et al. China Consortium for the Genetics of Autoimmune Thyroid Disease. Robust evidence for five new Graves’ disease risk loci from a staged genome-wide association analysis. Hum Mol Genet 2013; 22:3347.
79. Tomer Y, Hasham A, Davies T et al. Fine mapping of loci linked to autoimmune thyroid disease identifies novel susceptibility genes. J Clin Endocrinol Metab 2013; 98:E144.
80. Kutluturk F, Yarman S, Sarvan F et al. Association of Cytokine Gene Polymorphisms (IL6, IL10, TNF-α, TGF-β and IFN-γ) and Graves’ Disease in Turkish Population. Endocr Metab Immune Disord Drug Targets 2013; 13:163.
81. Guo T, Huo Y, Zhu W et al. Genetic association between IL-17F gene polymorphisms and the pathogenesis of Graves’ Disease in the Han Chinese population. Gene 2013; 512:300.
82. Feng M, Li H, Chen S et al. Polymorphisms in the vitamin D receptor gene and risk of autoimmune thyroid diseases: a meta-analysis.Endocrine 2013; 43:318.
83. Inoue N, Watanabe M, Yamada H et al. Associations between autoimmune thyroid disease prognosis and functional polymorphisms of susceptibility genes, CTLA4, PTPN22, CD40, FCRL3, and ZFAT, previously revealed in genome-wide association studies. J Clin Immunol 2012; 32:1252.
84. Alkhateeb A, Jarun Y & Tashtoush R. Polymorphisms in NLRP1 gene and susceptibility to autoimmune thyroid disease. Autoimmunity 2013; 46:215.
85. Vural P, Baki M, Dogru-Abbasoglu S et al. Vascular endothelial growth factor polymorphisms increase the risk of developing Graves’ disease. Int Immunopharmacol 2012; 14:132.
86. Arakawa Y, Watanabe M, Inoue N et al. Association of polymorphisms in DNMT1, DNMT3A, DNMT3B, MTHFR and MTRR genes with global DNA methylation levels and prognosis of autoimmune thyroid disease. Clin Exp Immunol 2012; 170:194.
87. Morita M, Watanabe M, Inoue N et al. Functional polymorphisms in TBX21 and HLX are associated with development and prognosis of Graves’ disease. Autoimmunity 2012; 45:129.
88. Prummel M, Wiersinga W. Smoking and risk of Graves’ disease. JAMA 1993; 269: 479.
89. Rasooly L, Burek C, Rose N. Iodine-induced autoimmune thyroiditis in NOD-H-2h4 mice. Clin Immunol Immunopathol 1996; 81:287.
90. Falgarone G, Heshmati, Cohen R et al. Role of emotional stress in the pathophysiology of Graves’ disease. Eur J Endocrinology 2013; 168: R13.
91. Baker JR Jr. Autoimmune endocrine disease. JAMA 1997; 278: 1931.
92. Mori K, Nakagawa Y, Ozaki H. Does the gut microbiota trigger Hashimoto’s thyroiditis? Discov Med 2012; 14:321.
93. Zois C, Stavrou I, Kalogera C et al. High prevalence of autoimmune thyroiditis in schoolchildren after elimination of iodine deficiency in northwestern Greece. Thyroid 2003; 13: 485.
94. Saboori A, Rose N, Bresler H et al. Iodination of human thyroglobulin (Tg) alters its immunoreactivity. I: Iodination alters multiple epitopes of human Tg. Clin Exp Immunol 1998; 113: 297.
95. Vitale M, Di Matola T, D’Ascoli F et al. Iodide excess induces apoptosis in thyroid cells through a p53-independent mechanism involving oxidative stress. Endocrinology 2000; 141: 598.
96. Fountoulakis S, Philippou G, Tsatsoulis A. The role of iodine in the evolution of thyroid disease in Greece: from endemic goiter to thyroid autoimmunity. Hormones (Athens) 2007; 6:25.
97. Yu X, Li L, Li Q et al. TRAIL and DR5 promote thyroid follicular cell apoptosis in iodine excess-induced experimental autoimmune thyroiditis in NOD mice. Biol Trace Elem Res 2011; 143:1064.
98. Nakahara M, Nagayama Y, Saitoh O, Sogawa R, Tone S, Abiru N. Expression of immunoregulatory molecules by thyrocytes protects nonobese diabetic-H2h4 mice from developing autoimmune thyroiditis. Endocrinology 2009; 150:1545.
99. Wang SH, Cao Z, Wolf JM, Van Antwerp M, Baker JR Jr. Death ligand tumor necrosis factor-related apoptosis-inducing ligand inhibits experimental autoimmune thyroiditis. Endocrinology 2005; 146:4721.
100. Bagnasco M, Bossert I, Pesce G Stress and autoimmune thyroid diseases. Neuroimmunomodulation 2006; 13: 309.
101. Winsa B, Karlsson A, Bergstrom R et al. Stressful life events and Graves’ disease. Lancet 1991; 338: 1475.
102. Paunkovic N, Paunkovic J, Pavlovic O et al. The significant increase in incidence of Graves’ disease in eastern Serbia during the civil war in the former Yugoslavia (1992 to 1995). Thyroid 1998; 8: 37.
103. Tsatsoulis A. The role of stress in the clinical expression of thyroid autoimmunity. Ann N York Acad Sc 2006; 1088: 382.
104. Kim BJ, Jones HP. Epinephrine-primed murine bone marrowderived dendritic cells facilitate production of IL-17A and IL-4 but not IFN-g by CD4CT cells. Brain, Behavior, and Immunity 2010; 24: 1126.
105. Vgontzas AN, Zoumakis E, Bixler E et al. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J Clin Endocrinol and Metab 2004; 89:2119.
106. Miller G, Chen E, Sze J et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kB signaling. Biological Psychiatry 2008; 64:266.
107. Benvenga S. Benzodiazepine and remission of Graves’ disease. Thyroid 1996; 6: 659.
108. Krysiak R, Okopien B. The effect of levothyroxine and selenomethionine on lymphocyte and monocyte cytokine release in women with Hashimoto’s thyroiditis. J Clin Endocrinol Metab 2011; 96:2206.
109. Xue H, Wang W, Li Y et al. Selenium upregulates CD4(+)CD25(+) regulatory T cells in iodine-induced autoimmune thyroiditis model of NOD.H-2(h4) mice. Endocr J 2010; 7:595.
110. Duntas L, Mantzou E, Koutras D. Effects of a six month treatment with selenomethionine in patients with autoimmune thyroiditis. Eur J Endocrinol 2003; 148:389.
111. Mazokopakis E, Papadakis J, Papadomanolaki M et al. Effects of 12 months treatment with L-selenomethionine on serum anti-TPO Levels in Patients with Hashimoto’s thyroiditis. Thyroid 2007; 17:609.
112. Toulis et al. Selenium supplementation in the treatment of HT: a systematic review and a meta-analysis.Thyroid 2010; 20: 1163.
113. Liu R, Ma X, Xu L et al. Differential MicroRNA Expression in Peripheral Blood Mononuclear Cells from Graves’ Disease Patients. J Clin Endocrinol Metab 2012; 97:E968.
114. Zhou H, Huang X, Cui H et al. miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood 2010; 116:5885.
115. Guo L, Lu Z. The fate of miRNA* strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? PLoS One 2010; 5:e11387.
116. Cogni G, Chiovato L. An overview of the pathogenesis of thyroid autoimmunity HORMONES (Athens) 2013; 12:19.
117. Fugazzola L, Cirello V, Beck-Peccoz P. Microchimerism and Endocrine Disorders. J Clin Endocrinol Metab 2012; 97:1452.
118. O’Donoghue K, Chan J, de la Fuente J et al . Microchimerism in female bone marrow and bone decades after fetal mesenchymal stem-cell trafficking in pregnancy. Lancet 2004; 364:179.
119. Khosrotehrani K, Bianchi D. Fetal cell microchimerism: helpful or harmful to the parous woman? Curr Opin Obstet Gynecol 2003; 15:195.
120. Khosrotehrani K, Bianchi D. Multi-lineage potential of fetal cells in maternal tissue: a legacy in reverse. J Cell Sci 2005; 118:1559.
121. Sunami R, Komuro M, Yuminamochi T et al. Fetal cell microchimerism develops through the migration of fetus derived cells to the maternal organs early after implantation. J Reprod Immunol 2010; 84:117.
122. Fugazola L, Cirello V, Beck-Peccoz P et al. Microchimerism and endocrine disorders. J Clin Endocrinol Metab 2012; 97:1452.
123. Srivatsa B, Srivatsa S, Johnson K et al. Microchimerism of presumed fetal origin in thyroid specimens from women: a case-control study. Lancet 2001; 358:2034.
124. Friedrich N, Schwarz S, Thonack J et al. Association between parity and autoimmune thyroiditis in a general female population. Autoimmunity 2008; 41:174.
125. Bulow Pedersen I, Laurberg P, Knudsen N et al. Lack of association between thyroid autoantibodies and parity in a population study argues against microchimerism as trigger of thyroid autoimmunity. Eur J Endocrinol 2006; 154: 39.
126. Sgarbi J, Kasamatsu T, Matsumura L et al. Parity is not related to autoimmune thyroid disease in a population-based study of Japanese-Brazilians. Thyroid 2010; 20: 1151.
127. Yu S et al. B-cell deficient NOD.H-2h4 mice have CD4+CD25+ T regulatory cells that inhibit the development of spontaneous autoimmune thyroiditis. J Exp Med 2006; 203:349.
128. Saitoh O, Nagayama Y. Regulation of Graves’ hyperthyroidism with naturally occurring CD4+CD25+ regulatory T cells in a mouse model. Endocrinology 2006; 147:2417.
129. Morris GP, Kong YC. Tolerance to autoimmune thyroiditis: (CD4+)CD25+ regulatory T cells influence susceptibility but do not supersede MHC class II restriction. Front Biosci 2006; 11:1234.
130. Gangi E et al. IL-10-producing CD4+CD25+ regulatory T cells play a critical role in granulocyte-macrophage colony-stimulating factor-induced suppression of experimental autoimmune thyroiditis. J Immunol 2005; 174:7006.
131. Vasu C et al. Selective induction of dendritic cells using granulocyte macrophage-colony stimulating factor, but not fms-like tyrosine kinase receptor 3-ligand, activates thyroglobulin-specific CD4+/CD25+ T cells and suppresses experimental autoimmune thyroiditis. J Immunol 2003; 170:5511.
132. Bossowski A, Moniuszko M, Dabrowska M et al. Lower proportions of CD4+CD25(high) and CD4+FoxP3, but not CD4+CD25+CD127(low) FoxP3+ T cell levels in children with autoimmune thyroid diseases. Autoimmunity 2013; 46:222.
133. Figueroa-Vega N, Alfonso-Pe/rez M, Benedicto I et al. Increased circulating proinflammatory cytokines and Th17 lymphocytes in Hashimoto’s thyroiditis. J Clin Endocrinol and Metab 2010; 95: 953.
134. Glick AB, Wodzinski A, Fu P et al. Impairment of regulatory T-cell function in autoimmune thyroid disease. Thyroid 2013; 23:871.
135. Pan D, Shin YH, Gopalakrishnan G, Hennessey J, De Groot LJ. Regulatory T cells in Graves’ disease. Clin Endocrinol (Oxf) 2009; 71:587.
136. Mao C, Wang S, Xiao Y et al. Impairment of regulatory capacity of CD4+CD25+ regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves’ disease. J Immunol 2011; 186:4734.
137. Hu et al, Tian W, Zhang L. Function of regulatory T-cells improved by dexamethasone in Graves disease. Eur J Endocrinol 2012; 166:641.
138. Nanba T, Watanabe M, Inoue N & Iwatani Y. Increases of the Th1/Th2 cell ratio in severe Hashimoto’s disease and in the proportion of Th17 cells in intractable Graves’ disease. Thyroid 2009; 19: 495.
139. Wang SH et al. A unique combination of inflammatory cytokines enhances apoptosis of thyroid follicular cells and transforms nondestructive to destructive thyroiditis in experimental autoimmune thyroiditis. J Immunol 2002; 168:2470.
140. Battifora M et al. B7.1 costimulatory molecule is expressed on thyroid follicular cells in Hashimoto’s thyroiditis, but not in Graves’ disease. J Clin Endocrinol Metab 1998; 83:4130.
141. Mazziotti G et al. Type-1 response in peripheral CD4+ and CD8+ T cells from patients with Hashimoto’s thyroiditis. Eur J Endocrinol 2003; 148:383.
142. Nagayama Y, Nakahara M, Shimamura M et al. Prophylactic and therapeutic efficacies of a selective inhibitor of the immunoproteasome for Hashimoto’s thyroiditis, but not for Graves’ hyperthyroidism, in mice. Clin and Experiment Immunol 2012; 168: 268.
143. Stassi G et al. Control of target cell survival in thyroid autoimmunity by T helper cytokines via regulation of apoptotic proteins. Nat Immunol 2000; 1:483.
144. Yamamoto K, Itoh M, Okamura T et al. Relative levels of the inflammatory cytokine TNFα and the soluble CD40 ligand profile in serum correlate with the thyrotoxic activity of Graves’ disease. Thyroid 2012; 22:516.
145. Coles A et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet 1999; 354:1691.
146. Li X, Qi Y, Ma X et al. Chemokine (C-C Motif) Ligand 20, a Potential Biomarker for Graves’ Disease, Is Regulated by Osteopontin PLoS One 2013; 8:e64277.
147. Zheng L, Ye P& Liu C. The role of the IL-23/IL-17 axis in the pathogenesis of Graves’disease Endocrine Journal 2013; 60: 591.
148. Tsai K, Tsai F, Lin H et al. Thymic stromal lymphopoietin gene promoter polymorphisms and expression levels in Graves’ disease and Graves’ ophthalmopathy BMC Medical Genetics 2012; 13:116.
149. He R, Geha R. Thymic stromal lymphopoietin. Ann N Y Acad Sci 2010; 1183:13.
150. Tanaka J, Watanabe N, Kido M et al. Human TSLP and TLR3 ligands promote differentiation of Th17 cells with a central memory phenotype under Th2-polarizing conditions. Clin Exp Allergy 2009; 39:89.
151. Antonelli A, Ferrari SM, Corrado A, Ferrannini E, Fallahi P. Increase of Interferon-γ Inducible CXCL9 and CXCL11 Serum Levels in Patients with Active Graves’ Disease and Modulation by Methimazole Therapy. Thyroid 2013; 23:1461.
152. O’Regan A, Hayden J & Berman J. Osteopontin augments CD3- mediated interferon-gamma and CD40 ligand expression by T cells, which results in IL-12 production from peripheral blood mononuclear cells. J Leukoc Biol 2000; 68: 495.
153. Xu L, Ma X, Wang Y et al. The expression and pathophysiological role of osteopontin in Graves’ disease. J Clin Endocrinol Metab 2011; 96:E1866.70.
154. Qi Y, Li X, Ma X et al. The role of osteopontin in the induction of the CD40 ligand in Graves’ disease. Clin Endocrinol (Oxf) 2013; doi: 10.1111/cen.12229. (Epub ahead of print).
155. Li J, Hong F, Zeng J, Huang G. Functional interleukin-17 receptor A are present in the thyroid gland in intractable Graves disease. Cell Immunol 2013; 28:85.
156. da Rocha Jr L, Duarte L, Dantas A et al. Increased serum interleukin 22 in patients with rheumatoid arthritis and correlation with disease activity. J Rheumatol 2012; 39:1320.
157. Pan H, Li X, Zheng S, Ye D. Emerging role of interleukin-22 in autoimmune diseases. Cytokine Growth Factor Rev 2013; 24:51.
158. Semerano L, Assier E, Boissier M. Anti-cytokine vaccination: a new biotherapy of autoimmunity? Autoimmun Rev 2012; 11:785.
159. Peng D et al. A high frequency of circulating th22 and th17 cells in patients with new onset graves’ disease. Plos One 2013; 8: e68446.
160. Zha B, Wang L, Liu X et al. Decrease in proportion of CD19+ CD24(hi) CD27+ B cells and impairment of their suppressive function in Graves’ disease. PLoS One. 2012; 7:e49835.
161. Vannucchi G, Covelli D, Curro N et al. Serum BAFF concentrations in patients with Graves’ disease and orbitopathy before and after immunosuppressive therapy. J Clin Endocrinol Metab 2012; 97:E755.
162. Wang F, Yan T, Chen L et al. Involvement of inducible costimulator ligand (ICOSL) expression in thyroid tissue in hyperthyroidism of Graves’ disease patients. J Clin Immunol 2012; 32:1253.
163. Weetman AP. Cellular immune responses in autoimmune thyroid disease. Clin Endocrinol (Oxf) 2004; 61:405.
164. Kemp EH et al. Detection and localization of chemokine gene expression in autoimmune thyroid disease. Clin Endocrinol (Oxf) 2003; 59:207.
165. Armengol M, Juan M, Lucas-Martin A et al. Demonstartion of thyroid antigen specific B cells and recombination-activating gene expression in chemokine–containing active inthrarthyroidal germinal centers. Am J Pathol 2001; 159:861.
166. Ruwhof C, Drexhage HA. Iodine and thyroid autoimmune disease in animal models. Thyroid 2001; 11: 427.
167. Afzali B, Lombardi G, Lechler R, Lord G. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol 2007; 148: 32.
168. Todd I, Pujol-Borrell R, Hammond L et al. Interferon-gamma induces HLA-DR expression by thyroid epithelium. Clin Exp Immunol 1985; 61: 265.
169. Fountoulakis S, Vartholomatos G, Kolaitis N et al. Differential expression of Fas system apoptotic molecules in peripheral lymphocytes from patients with Graves’ disease and Hashimoto’s thyroiditis. Eur J Endocrinol 2008; 158:853.
170. Andrikoula M, Tsatsoulis A. The role of Fas-mediated apoptosis in thyroid disease. Eur J Endocrinol 2001; 144: 561.
171. Tsatsoulis A. The role of apoptosis in thyroid disease. Minerva Medica 2002; 93: 169.
172. Cheng J, Zhou T, Liu C et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 1994; 263: 1759.
173. Martinez-Lorenzo M, Alava M, Anel A et al. Release of preformed Fas ligand in soluble form is the major factor for activation-induced death of Jurkat T cells. Immunology 1996; 89: 511.
174. Mitsiades N, Poulaki V, Kotoula V et al. Fas/Fas ligand up-regulation and Bcl-2 down regulation may be significant in the pathogenesis of Hashimoto’s thyroiditis. J Clin Endocrinol Metab 1998; 83: 2199.
175. Giordano C, Richiusa P, Bagnasco M et al. Differential regulation of Fas-mediated apoptosis in both thyrocyte and lymphocyte cellular compartments correlates with opposite phenotypic manifestations of autoimmune thyroid disease. Thyroid 2001; 11: 233.
176. Andrikoula M, Vartholomatos G, Tsangaris G et al. Fas and Bcl-2 protein expression in thyrocytes of patients with nodular goiter. Eur J Endocrinol 2001; 145: 403.
177. Di Matola T, Mueller F, Fenzi G et al. Serum withdrawal-induced apoptosis in thyroid cells is caused by loss of fibronectin-integrin interaction. J Clin Endocrinol Metab 2000; 85: 1188.
178. Roura-Mir C, Catalfamo M, Sospedra M et al. Single-cell analysis of intrathyroidal lymphocytes shows differential cytokine expression in Hashimoto’s and Graves’ disease. Eur J Immunol 1997; 27: 3290.
179. Brown R, Bellisario R, Botero D et al. Incidence of transient congenital hypothyroidism due to maternal thyrotropin receptor-blocking antibodies in over one million babies. J Clin Endocrinol Metab 1996; 81: 1147.
Created: October 25, 2014
Last update: October 25, 2014