
Χαράλαμπος Λυσίκατος
Κωνσταντίνος Α. Στρατάκης
Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), NIH
1. Εισαγωγή
Στην Ιατρική ο όρος μελαγχρωματικά στίγματα ή κηλίδες προέρχεται από τη λατινική λέξη lentigo (που σημαίνει μικρή φακή) και αποτελούν επίπεδα χρωσμένες κηλίδες στο δέρμα και στο επιθήλιο1. Χαρακτηρίζονται από το μικρό τους μέγεθος (<0.5 cm), ακανόνιστα όρια και διακριτές σημάνσεις από διαφορετικές χρώσεις καφέ και μαύρο. Τα οικογενή σύνδρομα μελαγχρωματικών στιγμάτων ή κηλίδων σχετίζονται με αυξημένη συχνότητα νευρικών, ενδοκρινολογικών και μεσεγχυματικών όγκων2. Η ιστολογική εξέταση αυτών αποκαλύπτει πάχυνση της επιδερμίδας, υπέρχρωση των κυττάρων της βασικής στιβάδας και υπερπλασία των μελανοκυττάρων. Το τελευταίο χαρακτηριστικό βοηθά στην ιστολογική διαφορική διάγνωση από τις εφελίδες, οι οποίες περιέχουν φυσιολογικό αριθμό μελανοκυττάρων και είναι χρωσμένες, λόγω της αυξημένης μελανίνης στα κερατινικά κύτταρα της βασικής στιβάδας (Εικόνα 1).
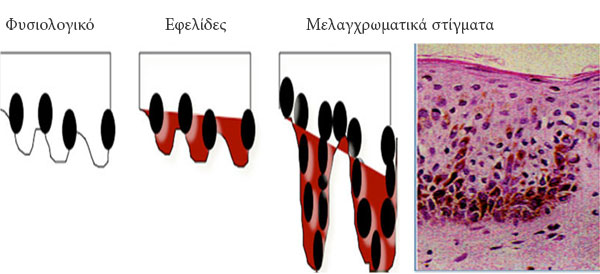
Εικόνα 1. Ιστολογική εικόνα των εφελίδων και των μελαγχρωματκών στιγμάτων. Η μεγένθυση των μελαγχρωματκών στιγμάτων (δεξιά) απεικονίζει τη χαρακτηριστική μελανοκυτταρική διόγκωση.
Οι εφελίδες απαντώται σχεδόν αποκλειστικά στις επιφάνειες του σώματος που εκτίθενται στον ήλιο, ενώ τα μελαγχρωματικά στίγματα σε οποιαδήποτε επιφάνεια του σώματος και σε αντίθεση με τις εφελίδες δε σκουραίνουν με τον ήλιο. Ιδιαίτερες περιοχές υπέρχωσης, όπως οι παλάμες, τα πέλματα, ο επιπεφυκότας, το όριο των χειλιών μεταξύ του βλεννογόνου μεταξύ και δέρματος, αποτελούν χαρακτηριστικά σημεία των μελαγχρωματικών στιγμάτων και παρέχουν σημαντικές ενδείξεις για την παρουσία υποκείμενου συνδρόμου ή συστηματικής νόσου1.
2. Σύνδρομο Peutz–Jeghers
Το σύνδρομο Peutz-Jeghers είναι το πιο γνωστό από τα σύνδρομα με μελαγχρωματικά στίγματα. Άλλα σύνδρομα είναι το σύμπλεγμα Carney (Carney complex, CNC), το σύνδρομο Laugier-Hunziker, το σύνδρομο Bannayan-Zonnana, η νόσος του Cowden και το σύνδρομο LEOPARD/Noonan, που θα περιγραφούν παρακάτω.
Το σύνδρομο Peutz-Jeghers (PJS) είναι ένα σπάνιο γενετικά σύνδρομο, που κληρονομείται με τον αυτοσωμικά επικρατούντα χαρακτήρα, συχνότητα 2.2/100.00 και επισύρει υψηλό ρίσκο για κακοήθεια. Η συσχέτιση μεταξύ των μελαγχρωματικών στιγμάτων και των αμαρτωματωδών πολυπόδων του γαστρεντερικού συστήματος έγινε από τους Dr. Peutz και Jeghers3,4. Το σύνδρομο χαρακτηρίζεται από αυξημένα ποσοστά όγκων, συμπεριλαμβανομένων όγκων του γαστρεντερικού , αποτιτανωμένους όγκους Sertoli των όρχεων, καρκίνο του τραχήλου της μήτρας, καρκίνο του μαστού, καρκίνο του παγκρέατος και καρκίνο του ενδομητρίου. Διαγνωστκά στοιχεία για το σύνδρομο Peutz-Jeghers αποτελούν οι αμαρτωματώδεις πολύποδες και η υπέρχρωση του βλεννογόνων. Η υπέρχρωση χαρακτηρίζεται από συνήθως σκούρες καφέ-μπλε χρώματος κηλίδες, οι οποίες πιο συχνά εντοπίζονται στα όρια των χειλιών, στο βλεννογόνο της στοματικής κοιλότητας και του γαστρενετερικού, όπως και στις παλάμες, τα πέλματα και την περιπρωκτική περιοχή5. Τα μελαγχρωματικά στίγματα συνήθως απαντώνται νωρίς κατά την παιδική ηλικία και έχουν την τάση να εξασθενούν με την ενηλικίωση. Σπανίως δε ενδείκνυται βιοψία.
Η πλειάδα των περιπτώσεων του συνδρόμου Peutz-Jeghers (PJS) οφείλονται σε ετερόζυγες μεταλλάξεις στη σερίνη-θρεονίνη κινάση STK11/LKB11, ένα γονίδιο καταστολέας ανάπτυξης όγκων, που βρίσκεται στο χρωμόσωμα 19p13.36,7. Μεταλλάξεις στο STK11/LKB11 γονίδιο βρίσκονται περίπου στο 80% των ασθενών με PJS. Το γονιδίο STK11 είναι ένα κλασσικό γονιδίο καταστολέας ανάπτυξης όγκων, όπως αποδεικνύεται από την απώλεια της ετεροζυγωτίας (LOH: Loss Of Heterozygosity) στα αμαρτώματα και στα αδενοκαρκινώματα των ασθενών με PJS. Οι γαστρεντερικοί πολύποδες στο σύνδρομο αναπτύσσονται μετά από γενετικές μεταλλάξεις στο STK11/LKB11 σε συνδυασμό με σωματική μετάλλαξη ή LOH του φυσιολογικού αλλίλιου8. Οι ασθενείς με PJS δεν παρουσιάζουν πιο αυξημένη συχνότητα δερματικού καρκίνου, αν και τα μελαγχρωματικά στίγματα είναι πιθανοί δερματολογικοί όγκοι9.
Το σύνδρομο PJS οφείλεται σε απορρύθμιση στην ανιούσα (upstream) οδό του mTOR (mammalian Target Of Rapamycin), καθώς η μετάλλαξη που οδηγεί σε απώλεια λειτουργίας του STK11/LKB11 γονιδίου αναστέλει την ενεργοποίηση της ΑΜΡ-πρωτεϊνικής κινάσης (ΑΜΡΚ), η οποία σηματοδοτεί (downstream) την κατιούσα οδό, για να αναστείλει το mTOR. Το mTOR είναι μέλος των φωσφοϊνοσιτιδο-3-κινάσεων και αποτελεί σημαντικό ρυθμιστή στην ανάπτυξη και τον πολλαπλασιασμό των κυττάρων10,11. Επίσης, έχει ρυθμιστικό ρόλο στη βιογένεση των ριβοσομάτων, στη μετάφραση των πρωτεϊνών και το σχηματισμό της ακτίνης στον σκελετό του κυττάρου12.
3. Σύμπλεγμα Carney
Το σύμπλεγμα Carney (Carney complex – CNC, OMIM## 160980, 608837): αποτελεί αυτοσωμική επικρατούσα πολλαπλή νεοπλασία και σύνδρομο μελαγχρωματικών στιγμάτων (lentiginosis sydrome) που χαρακτηρίζεται από κηλιδώδεις δερματικές αλλοιώσεις , μυξώματα στην καρδιά και σε άλλους ιστούς και διαφορετικού τύπου ενδοκρινείς όγκους. Οι τελευταίοι περιλαμβάνουν, την πρωτοπαθή χρωσμένη οζώδη νόσο του φλοιού των επινεφριδίων (primary pigmented nodular adrenocortical disease – PPNAD), υποφυσιακή υπερπλασία ή υποφυσιακοί όγκους (που χαρακτηρίζονται συχνότερα από υπερέκκριση αυξητικής ορμόνης και προλακτίνης), νεοπλασίες των γονάδων (μακροκυτταρικών αποτιτανωμένων όγκων των κυττάρων Sertoli ή των κυττάρων Leydig) και του θυρεοειδούς αδένος και σπανίως καλοήθη και κακόηθη μελανωτικά σβανώματα -psammomatous melanotic schwannomas13, 14. Tο CNC περιγράφηκε το 1985 από τον Παθολογοανατόμο Dr. Carney ως το σύμπλεγμα: μυξώματος, κηλιδoδών δερματικών υπερχρώσεων και ενδοκρινικής υπερδραστηριότητας15. Μέχρι σήμερα έχουν περιγραφεί περισσότερες από 600 ξεχωριστές περιπτώσεις του συμπλέγματος. Λόγω της συμμετοχής διαφορετικών ενδοκρινών αδένων, το CNC εμπιεριέχει χαρακτηριστικά με άλλες πολλαπλές ενδοκρινείς νεοπλασίες, όπως το σύνδρομο McCune Albright, την πολλαπλή ενδοκρινή νεοπλασία τύπου 1 και2 (MEN1 και MEN2, αντιστοίχως) και λόγω των δερματικών εκδηλώσεων, παρουσιάζει ομοιότητες με το PJS.
Αρχικά, το 1996 μια μελέτη γενετικής σύνδεσης (genetic linkage study), προσδιόρισε ως γενετικό τόπο (locus) για το CNC στο χρωμόσωμα 2p16 (αργότερα γνωστό ως CNC2 locus), κοντά στο γονίδιο που κωδικοποιεί το POMC (proopiomelanocortin)16. To 2000, αναφέρθηκαν οι πρώτες βλαστικές (germline) μεταλλάξεις στο PRKAR1A γονίδιο στο χρωμόσωμα 17q22-24 (γνωστό ως CNC1 locus) που κωδικοποιεί τη ρυθμιστική υπο-ομάδα τύπου 1-α της cAMP, της ενεργοποιημένης πρωτεϊνικής κινάσης Α (ΡΚ Α)17. Σήμερα, γνωρίζουμε ότι το PRKAR1A γονίδιο είναι υπεύθυνο για περισσσότερες από το 70% των μεταλλάξεων στους ασθενείς με CNC.
Η ΡΚΑ είναι ο κύριος ρυθμιστής των cAMP επιδράσεων στα ευκαρυωτικά κύτταρα. Στην ανενεργής της μορφή, η ΡΚΑ είναι ένας τετραμερές που συναθροίζεται από δυο διμερή – δυο ρυθμιστικών υπομονάδων και δυο καταλυτικών υπομονάδων (εινκόνα 2)18,19. Οι καταλυτικές υπομονάδες είναι κινάσεις σερίνης-θρεονίνης, ενώ οι ρυθμιστικές υπομονάδες καθορίζουν το μηχανισμό με τον οποίο, η cAMP μεταφράζει το εξωκυτταρικό σήμα σε βιολογική έκφραση20. Για να προσδεθούν στις καταλυτικές υπομονάδες, οι ρυθμιστικές υπόκεινται σε πλήρη διαμορφωτική αλλαγή, κατά την οποία δυο cAMP- προσδετικoί τόποι (domain), διαχωρίζονται και περιτυλίσσονατι γύρω από την καταλυτική υπομονάδα20 (εικόνα 2).
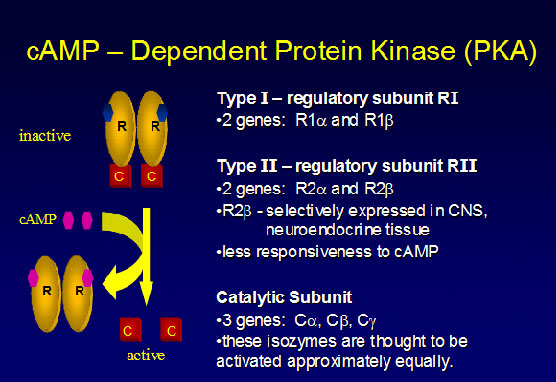
Εικόνα 2. cAMP – εξαρτώμενη πρωτεϊνική κινάση Α (ΡΚΑ). Αποτελείται από δυο διμερή – δυο ρυθμιστικών υπομονάδων (R – regulatory subunits) και δυο καταλυτικών υπομονάδων (C – catalytic subunits).
Με την αύξηση του ενδοκυτταρικού cAMP, δυο μόρια cAMP προσδένονται σε κάθε μια από τις ρυθμιστικές υπομονάδες, οδηγώντας σε αποσυναρμολόγηση του τετραμερούς στα ρυθμιστικά διμερή και τις καταλυτικές υπομονάδες. Οι ελεύθερες καταλυτικές υπομονάδες κατόπιν φωσφορυλίουν διαφορετικούς κυτταρικούς στόχους, ρυθμίζοντας τη μεταγραφή γονιδίων που μεσολαβούν στην κυτταρική ανάπτυξη, πολλαπλασιασμό και διαφοροποίηση21,22.
Ως τώρα, τέσσερις διαφορετικές ρυθμιστικές υπομονάδες: RΙα, RIβ, RIIα και RIIβ και τέσσερις καταλυτικές υπομονάδες (Cα, Cβ, C γ και PRKX) έχουν περιγραφεί στο ανθρώπινο είδος (εικόνα 2). Από όλες τις ΡΚΑ υπομονάδες, η PRKAR1A είναι η μόνη, στην οποία μεταλλάξεις είναι γνωστό ότι προκαλούν νόσο στο ανθρώπινο είδος23.
Το PRKAR1A γονίδιο βρίσκεται στο χρωμόσωμα 17q22-24 και αποτελείται από 11 εξόνια, 10 από τα οποία (2-11) κωδικοποιούν. Μετά την αναγνώριση του PRKAR1A ως το γονίδιο που προκαλεί το CNC, περισσότερες από 120 διαφορετικές αιτιολογικές μεταλλάξεις έχουν περιγραφεί. Οι γενετικές μεταλλάξεις στο CNC περιλαμβάνουν αντικατάσταση μια βάσης, μικρές ελλείψεις (deletions) μέχρι 15 bp ή συνδυασμένες ανακατατάξεις (combined rearrangements) κατά μήκος ολόκληρου του ανοικτού πλαισίου ανάγνωσης (open reading frame, OPF) του γονιδίου24.
Ο μηχανισμός με τον οποίο οι μεταλλάξεις στο PRKAR1A γονίδιο οδηγούν στο CNC οφείλεται σε μη αποτελεσματικό ελέγχο των ΡΚΑ καταλυτικών υπομονάδων, είτε λόγω απλοανεπάρκειας, είτε λόγω παραγωγής ελαττωματικής πρωτεΐνης. Ο μεγαλύτερος αριθμός των μεταλλάξεων στο PRKAR1A γονίδιο είναι απενεργοποιητικές – οδηγούν στη δημιουργία πρόωρου κωδικονίου λήξης και ακόλουθη διάσπαση (degradation) του μεταλλαγμένου mRNA, από ανερμηνεύσιμη καθοριζόμενη φθορά του mRNA (nonsense mediated mRNA decay)25,26. Oφείλονται σε ανερμηνεύσιμες αντικαταστάσεις (nonsense substitutions), μικρές ενθέσεις και διαγραφές (small insertions and deletions: INDELs) που οδηγούν σε μετατόπιση του αναγνωστικού πλαισίου και παραλλαγές διάσπασης (splice variants), όλες εντοπιζόμενες στον τελευταίο εξώνιο του PRKAR1A γονίδιου25. Oι φαινότυποι που οφείλονται σε πρόωρα κωδικόνια λήξης, ποικίλουν στις εκδηλώσεις τους και στη δριμύτητά της του κλινικά.
Άλλες μεταλλάξεις, είναι αυτές που οδηγούν σε επιμήκυνση της RIα πρωτεΐνης – τέσσερις μεταλλάξεις έχουν ως τώρα περιγραφεί, όλες εντοπιζόμενες στο τελευταίο εξώνιο του PRKAR1A γονιδίου και είναι κυρίως μικρές INDELs που οδηγούν σε αναδιάταξη του ανοικτού πλαισίου ανάγνωσης27.
Ένας σχετικός μικρός αριθμός των μεταλλάξεων στο PRKAR1A γονίδιο είναι αποτέλεσμα έκφρασης αλλοιωμένης (altered) πρωτεΐνης, λόγω παρερμηνεύσιμων αντικατάστασεων (missense substitutions), βραχέων INDELs και παραλλαγές διάσπασης.
Mεγάλες διαγραφές στην περιοχή του PRKAR1A γονιδίου (17q24.2-q24.3) έχουν περιγραφεί, σε ασθενείς με CNC, οι οποίοι ήταν αρνητικοί για μεταλλάξεις στο PRKAR1A γονίδιο στην ενζυματική μέθοδο αλληλουχίας Sanger. Αυτές οι περιπτώσεις χαρακτηρίζονται από εμφάνιση της κλινικής εικόνας σε νεότερη ηλικία, καθυστέρηση στην ανάπτυξη και σκελετικές ανωμαλίες28. Η τεχνολογία του συγκριτικού γενομικού υβριδισμού (array Comparative Genomic Hybridization – aCGH) έχει σημαντικό ρόλο στον καθορισμό αυτών των διαγραφών28.
Μετά την πρώτη περιγραφή της νόσου, ένας σημαντικός αριθμός ασθενών με CNC διαφορετικής εθνικότητας και με διαφορετικές κλινικές εκδηλώσεις έχουν αναφερθεί23. Τα πιο πρόσφατα διαγνωστικά κριτήρια για το CNC εμπεριέχονται στον παρακάτω πίνακα (πίνακα 1):
Πίνακας 1. Διαγνωστικά κριτήρια για το CNC.
| Μείζοντα Διαγνωστικά κριτήρια | |
| 1. | Κηλιδώδη δερματικά μελαγχρωματικά στίγματα με χαρακτηριστική διανομή (χείλη, επιπεφυκότα, έσω και έξω κανθούς, κολπικό βλεννογόνo και βλεννογόνο τους πέους) (Εικόνα 3) |
| 2. | Μύξωμα (δερματικό και στους βλεννογόνους) |
| 3. | Μύξωμα στην καρδιά |
| 4. | Μυξωμάτοση στο στήθος |
| 5. | Πρωτοπαθή χρωσμένη οζώδη νόσο του φλοιού των επινεφριδίων (PPNAD) ή παράδοξα θετική ανταπόκριση των γλυκορτικοστεροειδών στα ούρα, κατά τη χορήγηση δεξαμεθαζόνης (Liddle τεστ) |
| 6. | Γιγαντισμός ή ακρομεγαλία – εξαιτίας υποφυσιακού αδενώματος ή υπερπλασίας που παράγει αυξητική ορμόνη |
| 7. | Μακροκυτταρικoοί αποτιτανωμένοι όγκοι των κυττάρων Sertoli (Large cell calcifying Sertoli Cell Tumors – LCCSCT) ή χαρακτηριστικές αποτιτανώσεις στον υπέρηχο των όρχεων |
| 8. | Καρκίνο του θυροειδούς ή πολλαπλούς υποεχογενείς (ψυχρούς) όζους στον υπέρηχο του θυροειδούς αδένος, σε σχετικά νέους ασθενείς |
| 9. | Μελανωτικά σβανώματα – psammomatous melanotic schwannoma |
| 10. | Μπλε σπίλοι (πολλαπλοί) |
| 11. | Αδένωμα των πόρων (ductal adenoma) στο στήθος (συνήθως πολλαπλό) |
| 12. | Οστεοχονδρομύξωμα |
| Συμπληρωματικά κριτήρια | |
| 1. | Πρώτου βαθμού συγγενείς διαγνωσμένοι με τη νόσο |
| 2. | Απενεργοποιμένη μετάλλαξη στο PRKAR1A γονίδιο |
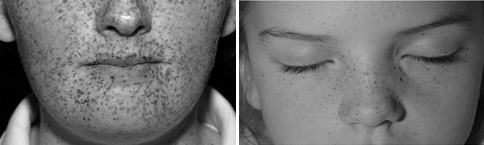
Εικόνα 3. Κηλιδώδη δερματικά μελαγχρωματικά στίγματα στα όρια χειλέων – βλεννογόνου και στους έσω κανθούς
Για να τεθεί η διάγνωση του CNC, είναι απαραίτητα δυο ή περισσότερα από τα μείζοντα διαγνωστικά κριτήρια. Οι μεταλλάξεις στο PRKAR1A γονίδιο μεταφέρονται κατά το αυτοσωμικό επικρατώντα μηχανισμό. Ιδιαιτέρως, σε ασθενείς με CNC που μελετήθηκαν με κλινική εικόνα συνδρόμου Cushing, 70 – 80% αυτών είχαν μεταλλάξεις στο PRKAR1A γονίδιο. Ανάμεσα στους φορείς των μεταλλάξεων, το σύνδρομο Cushing λόγω πρωτοπαθής χρωσμένης οζώδης νόσου του φλοιού των επινεφριδίων (PPNAD) συναντάται στο 70% των γυναικών με CNC πριν την ηλικία των 45 ετών, αλλά μόνο στο 45% των ανδρών με CNC, πιθανώς αντανακλώντας την υψηλότερη συχνότητα του συνδρόμου Cushing στο γυναικείο πληθυσμό29. Αυτά τα ποσοστά δεν υποδηλώνουν την ακριβή συχνότητα της PPNAD, καθ’ότι η συγκεκριμένη παθολογική εικόνα στα επινεφρίδια απαντάται σχεδόν στο 100% των ασθενών με CNC, στους οποίους έγινε αυτοψία30.
Η διεισδυτικότητα της νόσου στους ασθενείς με PRKAR1A μεταλλάξεις είναι πάνω από 95%. Συγκρίνοντας τους φορείς των PRKAR1A με ταλλάξεων, με ασθενείς που έχουν CNC χωρίς κάποια μετάλλαξη στο PRKAR1A γονίδιο, η νόσος παρουσιάζεται αργότερα στη διάρκεια της ζωής και αποτελεί συνήθως σποραδική περίπωση31. Οι ασθενείς χωρίς PRKAR1A μετάλλαξη μπορούν να ταξινομηθούν σε τρεις κατηγορίες: α) ασθενείς με τυπικές CNC εκδηλώσεις, β)ασθενείς με PPNAD και μελαγχρωματικά στίγματα και γ) ασθενείς με μεμονωμένη μικροοζώδη νόσο του φλοιού των επινεφριδίων (isolated Micronodular Adrenocortical Disease – iMAD).
Μια ευρεία έρευνα του γονιδιώματος των ασθενών με iMAD, οδήγησε στην αναγνώριση απενεργοποιητικών μεταλλάξεων στο γονίδιο που κωδικοποιεί τη φωσφοδιεστεράση (PDE) 11 A (PDE11A), το οποίο ακολουθήθηκε από την ανακάλυψη μεταλλάξεων σε έαν άλλο PDE γονίδιο – το PDE8Β.32,33 Και το PDE11A και το PDE8Β είναι cAMP ένζυμα διάσπασης και απενεργοποιητικές μεταλλάξεις οδηγούν, σε αύξηση του κυτταρικού επιπέδου του cAMP και ενεργοποίηση της ΡΚΑ σηματοδότησης, όπως αντίστοιχα συμβαίνει και στις ατέλειες (defects) του PRKAR1A στην ίδια οδό (pathway) σηματοδότησης.
Μελέτες που συσχετίζουν το γονότυπο με το φαινότυπο στους ασθενείς με CNC είναι περιορισμένες. Όλες οι μεταλλάξεις σχετίζονται με αυξημένη ΡΚΑ σηματόδοτηση, ως τον κύριο αίτιο για το σχηματισμό όγκων και τα άλλα συμπτώματα του συνδρόμου. Εντούτοις, μελέτες τα τελευταία 20 χρόνια ασθενών με CNC, έδειξαν συσχέτιση μεταξύ των μεταλλάξεων και των κλινικών εκδηλώσεων του συμπλέγματος. Συγκεντρωτικά, PRKAR1A μεταλλάξεις παρατηρήθηκαν στο 40% των ασθενών που ταξινομήθηκαν ως “σποραδικές” περιπτώσεις, σε αντίθεση με το περίπου 80% των ασθενών, στους οποίους βρέθηκε PRKAR1A μετάλλαξη. Οι υπο-ομάδες που επήλθαν περιλαμβάνουν: α) μεμονωμένες περιπττώσεις PPNAD, συνοδευόμενες μερικές φορές από μελαγχρωματικά στίγματα σε ασθενείς με ηλικία μικρότερη των 8 ετών –σπανίως φορείς κάποια μετάλλαξης στο PRKAR1A και σε ασθενείς με μεμονωμένη περιπττώση PPNAD και παρουσία κάποιας μετάλλαξης στο PRKAR1A (κυρίως της c.709-7_709-2del). Και β) ασθενείς με μυξώματα (στο δέρμα, την καρδιά ή σ το στήθος), καρκίνο του θυρεοειδούς αδένος και μακροκυτταρικών αποτιτανωμένων όγκων των κυττάρων Sertoli και σχετικά πιο συχνές PRKAR1A μεταλλάξεις.31
Αφότου τεθεί η διάγνωση του CNC, το screening πειλαμβάνει μαγνητική τομογραφία (MRI) της υπόφυσης για την ανίχνευση πιθανών υποφυσιακών αδενωμάτων, συνοδευόμενη από εργαστηριακό έλεγχο των επιπέδων αυξητικής ορμόνηνης (GH) και σωματομεδίνης – C (IGF-1), MRI της σπονδυλικής στήλης για την έγκαιρη ανίχνευση σβανωμάτων, υπέρηχο του θυρεοειδούς αδένος, υπέρηχο του όσχεου στους άνδρες και των ωοθηκών στις γυναίκες, μέτρηση του επιπέδου κορτιζόλης με συλλογή ούρων 24ωρου και στο αίμα (ημερήσια επίπεδα κορτιζόλης και αδρενοκορτικοτρόπου ορμόνης), ακολουθούμενα από τη δοκιμασία Liddle με δεξαμεθαζόνη για την επιβεβαίωση της PPNAD 23. Η δοκιμασία Liddle στη PPNAD, χαρακτηρίζεται από “παράδοξη αύξηση” του επιπέδου κορτιζόλης34 (Εικόνα 4).
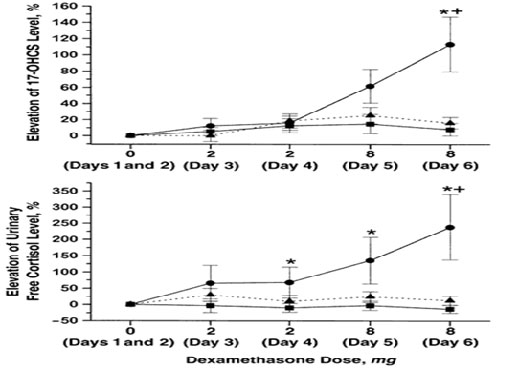
Εικόνα 4. κύκλοι: PPNAD (πρωτοπαθή χρωσμένη οζώδη νόσο του φλοιού των επινεφριδίων, τετράγωνα: MMAD (ογκώδη μακροοζώδη νόσο του φλοιού των επινεφριδίων) (Μassive Μacronodular Αdrenocortical Disease), τρίγωνα: μονήρη αδενώματα των επινεφριδίων
Το screening των ασθενών με CNC περιλαμβάνει επίσης υπέρηχο καρδιάς κάθε έξι μήνες στους ασθενείς με CNC που έχουν ιατρικό ιστορικό καρδιακών μυξωμάτων και ετησίως στους ασθενείς με CNC που έχουν αρνητικό ιστορικό καρδιακών μυξωμάτων. Η υποτροπή των καρδιακών μυξωμάτων είναι υψηλότερη στο γυναικείο πληθυσμό και μπορεί να οδηγήσει σε εγκεφαλικό επεισόδιο ως επιπλοκή, λόγω αποκόλλισης μικρών τμημάτων του μυξωμάτος και εισοδό τους στη συστηματική κυκλοφορία. Επομένως, η έγκαιρη ανίχνευση, τακτική παρακολούθση με υπέρηχο και MRI καρδιάς είναι πολύ σημαντικά στην αποτροπή των εγκαφακικών επεισοδίων35 (εικόνα 5).
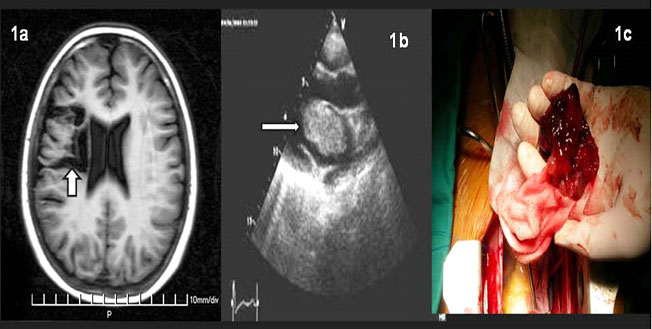
Εικόνα 5. a. ΜRI (T1-weighted) εγκεφάλου, περιοχή μειωμένης ένταση μαγνητικού σήματος (βέλος) εξαιτίας εγκεφαλομαλάκυνσης, απόροια ισχαιμικού επεισοδίου; b. Υπέρηχος καρδιάς , στον οποίο φαίνεται το καρδιακό μύξωμα στον αριστερό κόλπο (βέλος). c. Μακροσκοπική εικόνα του μυξώματος
4. Άλλα σύνδρομα
Άλλα σύνδρομα με με μελαγχρωματικά στίγματα και αμαρτώματα είναι το:
Σύνδρομο Laugier-Hunziker (LHS): μια σπάνια σποραδική διαταραχή, που περιγράφηκε το 1970 κια θα πρέπει να διαφορογιγνώσκεται από το PJS, λόγω της ομοιότητάς του στην παρουσία και τη διανομή των δερματικών βλαβών. Η διαφορά τους είναι ότι το LHS χαρακτηρίζεται στο 50-60% των ασθενών από υπέρχρωση (με καφέ ή σκουρόχρωμη χρώση) των νυχιών – μελανοονυχία – των παλάμων και των χειλιών, αλλά χωρίς τους πολύποδες στο γαστρεντερικό σύστημα36.
Το PTEN σύδρομο αμαρτοειδών όγκων, περιλαμβάνει το σύνδρομο Ruvalcaba-Myhre-Smith ή σύνδρομο Bannayan-Zonnana (ΒRRS), τη νόσο του Cowden, το PTEN σχετιζόμενο σύνδρομο Πρωτέα και το όμοιο του Πρωτέα σύνδρομο (Proteus-like syndrome): αποτελούν κληρονομικά μεταδιδόμενα νοσήματα που χαρακτηρίζονται από μεταλλάξεις στο PTEN (Phosphate και Tensin) γονίδιο (10q22-q23) στο 80% των ασθενών με νόσο του Cowden και 60% των ασθενών με ΒRRS. Το PTEN γονίδιο παίζει σημαντικό ρόλο στην κυτταρική ανάπτυξη, απόπτωση, διαφοροποίηση, και στην αλληλεπίδραση μεταξύ των κυττάρων37-40. Απώλεια της PTEN λειτουργίας οδηγεί σε ενεργοποίηση στη σηματοδότηση της πρωτεΐνης ΑΚΤ. Ένας από τους κατιούσης (downstream) στόχους της ΑΚΤ είναι το σύμπλεγμα tuberin-hamartin (TSC1/TSC2), μεταλλάξεις στο οποίο σχετίζονται με το αμαρτοματώδες σύνδρομο της οζώδης σκλήρυνσης41. Στην οζώδη σκήρυνση, απώλεια της ΡΤΕΝ λειτουργίας οδηγεί σε ενεργοπίηση της ΑΚΤ και κατιούσα ρύθμιση (down regulation) της tuberin/TSC2 και mTOR και επακόλουθη προαγωγή του κυτταρικού κύκλου και καταστολή της απόπτωσης. Η ανάκαλυψη αυτού του μονοπατιού, συνδέει της παθογένεση τους σχηματισμού όγκων στα αμαρτώδη όγκων σύνδρομα, όπως φαίνεται στο παρακάτω σχήμα (εικόνα 6).
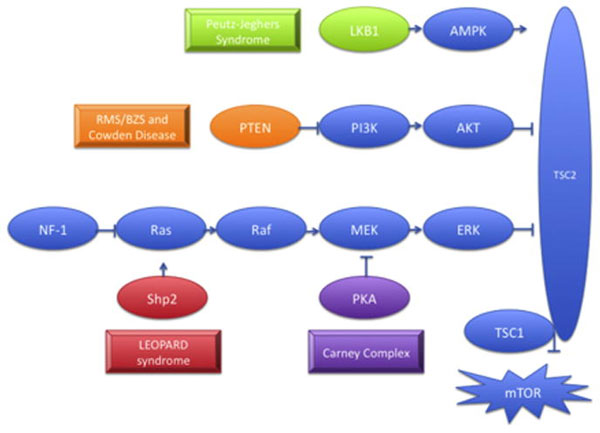
Εικόνα 6. Τα oγκοκατασταλτικά γονίδια LKB1, PTEN και PRKAR1A ασκούν ανασταλτικές δράσεις στη σηματοδότηση διαμέσου του TSC συμπλέγματος , που απενεργοποιεί το mTOR, ενώ η SHP2 ενίσχυση λειτουργίας (gain of function) μετάλλαξη οδηγεί σε κατιούσα mTOR ενεργοποίηση. Καθ’ένα από αυτά τα γονίδια είναι μεταλλαγμένο σε διακριτά σύνδρομα που χαρακτηρίζονται από την ανάπτυξη μελαγχρωματικών στιγμάτων και άλλων δερματικών βλαβών.
{Συντμήσεις: LKB1, serine/threonine kinase 11; AMPK, AMP-activated protein kinase; PTEN, phosphatase and tensin homolog; P13K, 1-phosphatidylinositol 3-kinase; AKT, protein kinase B; NF1; neurofibromatosis type 1MEK, mitogen activated protein kinase kinase; ERK, extracellular signal-regulated kinase; TSC1; tuberous sclerosis 1; TSC2; tuberous sclerosis 2; mTOR, mammalian target of rapamycin}
Περίπου 500 ασθενείς με παθογόνες γονιδιακές μεταλλάξεις στο PTEN έχουν αναφερθεί μέχρι τώρα42. Ασθενείς με PTEN μεταλλάξεις, έχουν αυξημένη πιθανότητα να αναπτύξουν πολλαπλά αμαρτώματα σε διάφορα όργανα, όπως το δέρμα, το στήθος, το θυροειδή αδένα, το κεντρικό νευρικό σύστημα και το γαστρεντερικό. Τα αμαρτώματα δεν είναι πραγματικοί όγκοι, αλλά αποτελόυν απόρεια συγκέντρωσης ιστών, που ανήκουν στο όργανο που βρέθηκε η συγκέντρωση αυτή, αλλά έχουν «λανθασμένα συναθροισθεί» στη διάρκεια της ανάπτυξης43.Το ΒRRS χαρακτηρίζεται από καθυστερημένη κινητική ανάπτυξη, μακροκεφαλία, λιπωμάτωση, καθώς και από την ύπαρξη μελαγχρωματικών κηλίδων στη βάλανο της πέους. Πρόσφατες μελέτες έχουν δείξει ότι το ΡΤΕΝ είναι ογκονίδιο κλειδί στη ανάπτυξη του βασικοκυτταρικού και εκ του πλακώδες επιθηλίου καρκίνου του δέρματος και στο μελάνωμα44, όπως και μεταλλάξεις αυτού ανευρίσκονται και στις περιπτώσεις αυτισμού45.
5. Σύνδρομο LEOPARD
LEOPARD είναι το ακρωνύμιο των:
Lentigines: μελαγχρωματικά στίγματα, Electrocardiographic conduction defects: διαταραχές αγωγιμότητας στο ηλεκτροκαρδιογράφημα, Ocular hypertelorism: Οφθαλμικός υπετελορισμός, Pulmonary stenosis: στένωση της πνευμονικής βαβίδας, Abnormal genitalia: ατελή ανάπτυξη των γεννητικών οργάνων, Retardation of growth: καθυστέρηση στην ανάπτυξη και sensorineural Deafness: νευροαισθητήρια κώφωση46
Η διάγνωση γίνεται από τα μελαγχρωματικά στίγματα σε συνδυασμό με δυο από τα άλλα χαρακτηριστικά. Τα μελαγχρωματικά στίγματα είναι συχνά η πρώτη κλινική εκδήλωση και απαντώνται στο πρόσωπο, στο άνω μέρος του κορμού, σπανιώς δε στο στοματικό βλεννογόνο , τα άκρα, τα εξωτερικά γεννητικά όργανα ή τον επιπεφυκότα47. Τα μελαγχρωματικά στίγματα στο σύνδρομο LEOPARD δεν περνούν το όριο μεταξύ δέρματος και βλεννογόνου στα χείλη, ένα χαρακτηριστικό που το διαφοροποιεί από το CNC και το PJS48.
Tα δυσμορφικά χαρακτηριστικά του συνδρόμου περιλαμβάνουν υπερτελορισμό και πτώση των βλεφάρων. Αυτά τα χαρακτηριστικά σε συνδυασμό με την αυξημένη συχνότητα της στένωσης της πνευμονικής βαλβίδας, έχουν σημαντική φαινοτυπική επικάλυψη με το σύνδρομο Noonan – μοριακά, τα δυο αυτά σύνδρομα, μοιράζονται το ίδιο αλλίλιο, PTPN11 γονίδιο στο χρωμόσωμα 12q24.149. Το PTPN11 κωδικοποιεί το SHP2 (εικόνα 3), ένα θετικό ρυθμιστή της RAS-MAPK σηματοδότησης. Μια ξεχωριστή μορφή του συνδρόμου LEOPARD οφείλεται σε RAF1 ανωμαλίες (χρωμόσωμα 3p25), αυτή η μορφή σχετίζεται με υπερτροφική καρδιομυοπάθεια50.
Τα μελαγχρωματικά στίγματα έχουν επίσης περιγραφεί και σε ασθενείς με εμφάνιση αρτηριακού διαχωρισμού (dissection) σε σχετικά νεαρή ηλικία, λόγω πιθανής ανωμαλίας σε κάποια κύτταρα που προέρχονται από τη νευρική ακρολοφία51.
Βιβλιογραφία
- Lodish MB, Stratakis CA (2011) The differential diagnosis of familial lentiginosis syndromes. Familial Cancer 10:481-490
- Fitzpatrick TB, Wolff K (2008) Fitzpatrick’s dermatology in general medicine, 7th edn. McGraw-Hill, New York
- Peutz JLA. Very remarkable case of familial polyposis of mucous membrane of intestinal tract and nasopharynx accompanied by peculiar pigmentations of skin and mucous membrane. (Dutch) Nederl Maandschr Geneesk. 1921;10:134–46
- Jeghers H, McKusick V, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241:993
- Tomlinson IP, Houlston RS (1997) Peutz-Jeghers syndrome. J Med Genet 34:1007–1011
- Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43
- Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–7
- Miyaki M, Iijima T, Hosono K, et al. Somatic mutations of LKB1 and beta-catenin genes in gastrointestinal polyps from patients with Peutz-Jeghers syndrome. Cancer Res. 2000;60:6311–3
- Rowan A, Bataille V, MacKie R et al. Somatic mutationsin the Peutz-Jeghers (LKB1/STKII) gene in sporadic malignant melanomas. J Invest Dermatol 1999;112:509–511
- Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(Spec No 2):R251–8
- Rosner M, Hanneder M, Siegel N, et al. The mTOR pathway and its role in human genetic diseases. Mutat Res. 2008;659:284–92
- Sandsmark DK, Pelletier C, Weber JD, et al. Mammalian target of rapamycin: master regulator of cell growthin the nervous system. Histol Histopathol 2007;22:895–903
- Carney, J A. The Carney complex (myxomas, spotty pigmentation, endocrine overactivity, and schwannomas). Dermatologic clinics 1995; 13: 19 -26
- Stratakis CA, Kirschner LS, Carney JA. Carney complex: diagnosis and management of the complex of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas. Am J Med Genet 1998; 80: 183-5
- Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985; 64:270-83
- Stratakis CA, Carney JA, Lin JP et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11kindreds and linkage to the short arm of chromosome 2. The Journal of Clinical Investigation 1996; 97: 699-705
- Bertherat J, Horvath A, Groussin L et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. Journal of Clinical Endocrinology and Metabolism 2009;94:2085-91.
- Kinderman FS, Kim C, von Daake S et al. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Molecular Cell 2006;24:397-408.
- Τasken K, Skalhegg BS, Tasken KA et al. Structure, function, and regulation of human cAMP-dependent protein kinases. Advances in Second Messenger and Phosphoprotein Research 1997; 31: 191-204.
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004; 155: 661-675
- Scott JD. Cyclic nucleotide-dependent protein kinases. Pharmacology & Therapeutics 1991; 50: 123-145
- Shabb JB. Physiological substrates of cAMP-dependent protein kinase. Chemical Reviews 2001; 101: 2381-2411
- Rothenbuhler A, Stratakis CA. Best Practice & Research Clinical Endocrinology & Metabolism 2010; 24: 389-399
- Horvath A, Bertherat J, Groussin L et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): An update. Human Mutation 2010; 4: 369-379
- Kirschner LS, Sandrini F, Monbo Jet al. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum Mol Genet. 2000; 9: 3037-3046
- Greene EL, Horvath AD, Nesterova M et al. In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum Mutat. 2008; 29: 633-639
- Patronas Y, Horvath A, Greene E, et al. In vitro studies of novel PRKAR1A mutants that extend the predicted RIα protein sequence into the 3′-untranslated open reading frame: proteasomal degradation leads to RIα haploinsufficiency and Carney complex. J Clin Endocrinol Metab. 2012; 97: E496-502
- Salpea P, Horvath A, London E, et al. Large deletions of the PRKAR1A locus at 17q24.2-q24.3 in Carney complex: genotype-phenotype correlations and implications for genetic testing J Clin Endocrinol Metab. 2013; on press
- Grumbach MM, Biller BM, Braunstein GD et al. Management of the clinically inapparent adrenal mass (“incidentaloma”). Ann Intern Med. 2003; 138: 424-429
- Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001; 86: 4041-4046
- Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 2009; 94: 2085-2091
- Gunther DF, Bourdeau I, Matyakhina L, et al. Cyclical Cushing syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease, or a new entity? J Clin Endocrinol Metab 2004; 89: 3173–3182
- Horvath A, Giatzakis C, Robinson-White A, et al. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res 2006; 66: 11571–11575
- Stratakis CA, Sarlis N, Kirschner LS, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Annals of Internal Medicine 1999; 131: 585-591
- Briassoulis G, Kuburovic V, Xekouki P, et al. Recurrent left atrial myxomas in Carney complex: a genetic cause of multiple strokes that can be prevented. Journal of stroke and cerebrovascular diseases. 2012; 21:914.e1-.e918.
- Lampe AK, Hampton PJ, Woodford-Richens K., et al. Laugier-Hunziker syndrome: an important differential diagnosis for Peutz-Jeghers syndrome. J Med Genet 2003; 40:e77
- Nelen MR, Padberg GW, Peeters EA et al. Localization of the gene for Cowden disease to chromosome 10q22–23. Nat Genet 1996; 13:114–116
- Nelen MR, van Staveren WC, Peeters EA et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet 1997; 6:1383–1387
- Marsh DJ, Coulon V, Lunetta KL et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan- Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet 1998; 7:507–515
- Gorlin RJ, Cohen MM Jr, Condon LM, Burke BA. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet 1992; 44:307–314
- Manning BD, Cantley LC (2003) United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans 31:573–578
- Tan MH, Mester J, Peterson C et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet 2011; 88:42–56
- Majno G, Joris I. Cells, tissue, and disease: principles of general pathology. Oxford University Press 2004, NY
- Ming M, He YY. PTEN: new insights into its regulation and function in skin cancer. J Invest Dermatol 2009; 129:2109–2112
- Lv JW, Cheng TL, Qiu ZL, et al. Role of the PTEN signaling pathway in autism spectrum disorder. Neurosci Bull 2013; http://www.neurosci.cn (DOI: 10.1007/s12264-013-1382-3)
- Gorlin RJ, Anderson RC, Blaw M. Multiple lentigenes syndrome. Am J Dis Child 1969; 117:652–662
- Abdelmalek NF, Gerber TL, Menter A. Cardiocutaneous syndromes and associations. J Am Acad Dermatol 2002; 46:161–183
- Bauer AJ, Stratakis CA. The lentiginoses: cutaneous markers of systemic disease and a window to new aspects of tumourigenesis. J Med Genet 2005; 42:801–810
- Pandit B, Sarkozy A, Pennacchio LA et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 2007; 39:1007–1012
- Razzaque MA, Nishizawa T, Komoike Y et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 2007; 39:1013–1017
- Schievink WI, Michels VV, Mokri B, et al. A familial syndrome of arterial dissections with lentiginosis. N Engl J Med 1995; 332:576-579
Created: February 25, 2015
Last update: February 25, 2015